
24.3: Técnicas Computacionales
- Page ID
- 54738
Esta sección se centrará en las técnicas de procesamiento de datos brutos del proyecto ENCODE. Antes de que se puedan analizar los datos ENCODE (por ejemplo, para el descubrimiento de motivos, análisis de coasociación, agregación de señales sobre elementos, etc.), los datos sin procesar deben procesarse.
Incluso antes de que se procesen los datos, se aplica algún control de calidad. El control de calidad es necesario por varias razones. Incluso sin anti-cuerpos, las lecturas no están uniformemente dispersas. Las razones biológicas incluyen fragmentación no uniforme del genoma, regiones de cromatina abiertas que se fragmentan más fácilmente y secuencias repetitivas sobrecolapsadas en genomas ensamblados. El proyecto ENCODE corrigió estos sesgos de varias maneras. Se eliminaron porciones del ADN antes de la etapa ChIP, eliminando grandes porciones de datos no deseados. También se realizaron experimentos de control sin el uso de anticuerpos. Finalmente, se utilizaron lecturas de secuencias de ADN de entrada de fragmentos como fondo.
Debido al ruido inherente en el proceso Chip-seq, algunas lecturas serán de menor calidad. Usando una métrica de calidad de lectura, se desecharon lecturas por debajo de un umbral.
Las lecturas más cortas (y en menor medida, lecturas más largas) pueden mapear exactamente a una ubicación (mapeo único), múltiples ubicaciones (mapeo repetitivo) o ninguna ubicación en absoluto (no mapeable) en el genoma. Hay muchas formas potenciales de lidiar con el mapeo repetitivo, que van desde la difusión probabilística de la lectura hasta el uso de un enfoque EM. Sin embargo, dado que el proyecto ENCODE pretende ser lo más correcto posible, no asigna lecturas repetitivas a ninguna ubicación.
Proyecto ENCODE. Todos los derechos reservados. Este contenido está exculeded de nuestro Creativo
Licencia Commons. Para obtener más información, consulte http://ocw.mit.edu/help/faq-fair-use/.
Si una muestra no contiene ADN suciente y/o si está sobresecuenciada, simplemente secuenciará repetidamente duplicados de PCR de un conjunto restringido de fragmentos de ADN distintos. Esto se conoce como una biblioteca de baja complejidad y no es deseable. Para resolver este problema, se crea un histograma con el número de duplicados y se desechan muestras con una fracción no redundante (NRF) baja.
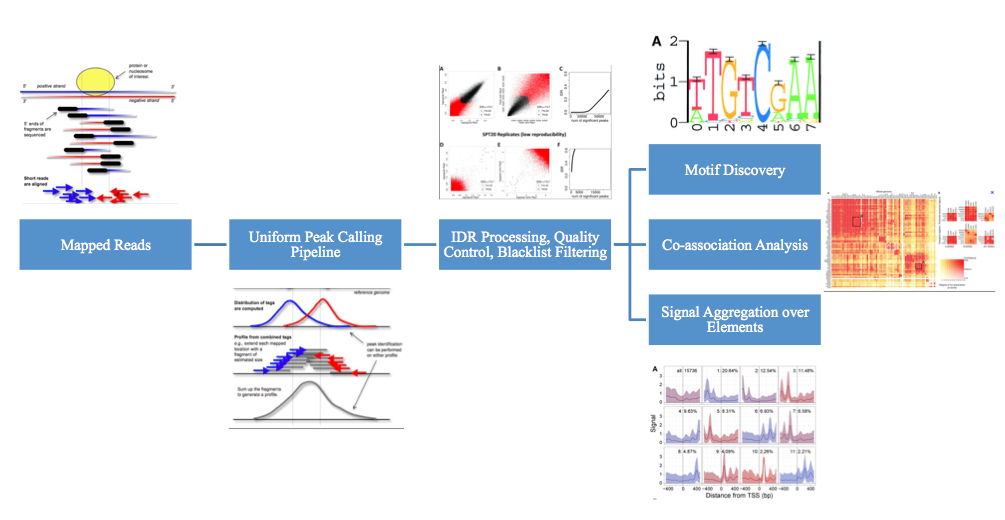
Chip-seq secuencias aleatorias de un extremo de cada fragmento, por lo que para determinar qué lecturas provienen de qué segmento, típicamente se usa el análisis de correlación cruzada de cadenas [Fig. 04]. Para lograr esto, se calculan las señales de hebra directa e inversa. Entonces, se desplazan secuencialmente el uno hacia el otro. En cada paso, se calcula la correlación. En el desplazamiento de longitud del fragmento f, los picos de correlación. f es la longitud a la que se fragmenta el ADN de ChIP. Mediante análisis adicionales, podemos determinar que debemos tener una correlación cruzada absoluta alta a la longitud del fragmento, y una correlación cruzada de longitud de fragmento alta en relación con la correlación cruzada de longitud de lectura. La RSC (Correlación Relativa de Strand) debe ser mayor a 1.
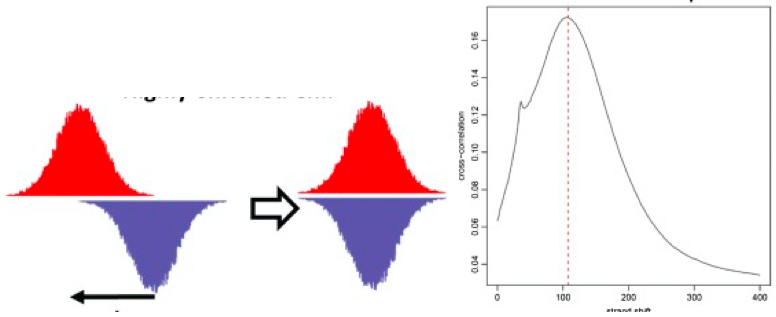
\[R S C=\frac{C C_{\text {fragment}}-\min (C C)}{C C_{\text {readlength}}-\min (C C)}\]
Una vez aplicado el control de calidad, los datos se procesan posteriormente para determinar las áreas reales de enriquecimiento. Para lograr esto, el proyecto ENCODE utilizó una versión modificada de peak calling. Existen muchos algoritmos de llamada de picos existentes, pero el proyecto ENCODE utilizó MACS y PeakSeq, ya que son deterministas. Sin embargo, no es posible establecer un valor p uniforme o una constante de tasa de descubrimiento falso (FDR). El FDR y el valor p dependen de ChIP y profundidad de secuenciación de entrada, la ubicuidad de unión del factor, y es altamente inestable. Además, diferentes herramientas requieren diferentes valores.
El proyecto ENCODE utiliza réplicas (del mismo experimento) y combina los datos para encontrar resultados más significativos. Las soluciones simples tienen grandes problemas: tomar la unión de los picos mantiene la basura de ambos, la intersección es demasiado estricta y arroja buenos picos, y tomar la suma de los datos no explota la independencia de los conjuntos de datos. En cambio, el proyecto ENCODE utiliza la tasa de descubrimiento independiente (IDR). La idea clave es que los verdaderos picos estarán altamente clasificados en ambas réplicas. Así, para encontrar picos significativos, los picos se consideran en orden de rango, hasta que los rangos ya no se correlacionan.
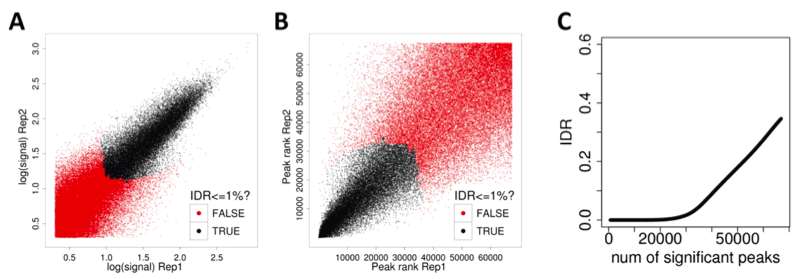
El corte podría ser diferente para las dos réplicas y los picos reales incluidos pueden diferir entre réplicas. Se modela como un modelo de mezcla gaussiana, que se puede ajustar a través de un algoritmo similar a EM. El uso de IDR conduce a una mayor consistencia entre las personas que llaman pico. Esto se debe a que la FDR solo se basa en el enriquecimiento sobre la entrada, IDR explota se replica. Además, usando métodos de muestreo, si solo hay una réplica, la tubería IDR aún se puede usar con pseudo-réplicas.