
30.6: Estudios de Asociación en todo el genoma
- Page ID
- 54975
En la década de 1990, los investigadores propusieron una metodología llamada asociación genómica para correlacionar sistemáticamente marcadores con rasgos. Estos estudios muestran grandes grupos de casos y controles, miden sus genotipos en el orden de un millón de marcadores e intentan correlacionar la variación (SNP, CNV, indeles) en sus genotipos con su variación en el fenotipo, rastreando la enfermedad a través de la población, en lugar de pedigríes.
Eventos Habilitando Estudios de Asociación en todo el genoma
Los estudios de asociación de todo el genoma (GWASs) son posibles debido a tres avances.
En primer lugar, los avances en nuestra comprensión del genoma y la creación de recursos genómicos nos han permitido comprender y catalogar mejor la variación del genoma. A partir de estos datos, nos hemos dado cuenta de la visión biológica clave de que los humanos son una de las especies con menor diversidad genética. Del orden de decenas de millones de SNP se comparten entre diferentes subpoblaciones humanas. Para cualquier región particular del genoma, observamos solo un número limitado de haplotipos (combinaciones de alelos que se heredan juntas). Esto se debe a que como especie, somos relativamente nuevos, y las mutaciones no han alcanzado nuestro rápido crecimiento. Debido a esta alta redundancia, solo necesitamos medir una fracción de todas las variantes en el genoma humano para capturarlas todas con LD. Luego podemos adaptar los algoritmos para inferir patrones de herencia en el análisis de ligamientos para imputar genotipos para los marcadores que no genotipo. Además, los recursos genómicos nos permiten elegir cuidadosamente marcadores para medir y hacer predicciones basadas en marcadores que muestran asociación estadísticamente significativa. Ahora tenemos la secuencia de referencia del genoma humano (permitiendo alineaciones, genotipos y llamadas SNP) y HapMap, un catálogo completo de SNP en humanos. También tenemos anotaciones genómicas de genes y elementos reguladores.
En segundo lugar, los avances en la tecnología de genotipado como los microarrays y la secuenciación de alto rendimiento nos han dado la oportunidad de comparar los genomas de los afectados con diversos fenotipos con los controles. También son los más fáciles y económicos de medir utilizando estas tecnologías. Aunque existen muchos tipos de variación en el genoma humano (la Figura 30.6 muestra algunos ejemplos), los SNP son la gran mayoría. Adicionalmente, para dar cuenta de los otros tipos de variantes, recientemente se han desarrollado microarrays de ADN para detectar la variación del número de copias además de los SNP, después de lo cual podemos imputar los datos no observados.
El tercer avance es una nueva expectativa de colaboración entre investigadores. Los GWAs se basan en tamaños de muestra grandes para aumentar la potencia (probabilidad de un verdadero positivo) de las pruebas estadísticas. La explosión en el número de GWASs publicados ha permitido un nuevo tipo de metaanálisis que combina los resultados de varios GWAs para el mismo fenotipo para hacer asociaciones más poderosas. El metaanálisis da cuenta de diversos sesgos técnicos y genéticos poblacionales en estudios individuales. Se espera que los investigadores que realizan GWASs colaboren con otros que han realizado GWAs en el mismo rasgo para mostrar la replicabilidad de los resultados. Al juntar los datos, también tenemos más confianza en las asociaciones reportadas, y los genes que se descubren pueden conducir al reconocimiento de vías y procesos clave.
¿Sabías?
Modificado del Wellcome Trust Sanger Institute: La enfermedad de Crohn y la Colitis Ulcerosa han sido focos para la genética de enfermedades complejas, y los esfuerzos masivos de colaboración del Consorcio Internacional de Genética de Enfermedades Inflamatorias Intestinales (IIBDGC) fortalecen el éxito de la investigación. Con aproximadamente 40,000 muestras de ADN de pacientes con EII y 20,000 controles sanos, el IIBDGC ha descubierto 99 loci definidos de IBD. En total, los 71 loci de la enfermedad de Crohn y 47 loci de CU representan el 23% y 16% de la heredabilidad de la enfermedad respectivamente. Los conocimientos clave sobre la biología de la enfermedad ya han resultado del descubrimiento de genes (por ejemplo, la autofagia en la enfermedad de Crohn, la función de barrera defectuosa en la UC y la señalización de IL23 en la EII y la enfermedad inmunomediada Se anticipa que de las muchas dianas farmacológicas novedosas identificadas por el descubrimiento de genes, algunas darán como resultado, en última instancia, una terapéutica mejorada para estas condiciones devastadoras. La mejora del diagnóstico, el pronóstico y la terapéutica son objetivos, con miras a una terapia personalizada (la práctica de usar el perfil genético de un individuo como guía para las decisiones de tratamiento) en el futuro.
Controles de Calidad
El principal problema en la realización de GWASs es eliminar los factores de confusión, pero las mejores prácticas se pueden usar para respaldar datos de calidad.
En primer lugar, existe el error de genotipado, que es lo suficientemente común como para requerir un tratamiento especial independientemente de la tecnología que se utilice. Este es un control técnico de calidad, y para dar cuenta de dichos errores, utilizamos umbrales en métricas como la frecuencia de alelos menores y la desviación del equilibrio Hardy-Weinberg y desechamos SNP que no cumplen con los criterios.
En segundo lugar, las diferencias genéticas sistemáticas entre subpoblaciones humanas requieren un control de calidad genética. Existen varios métodos para dar cuenta de esta subestructura poblacional, como el control genómico [7], pruebas de inconsistencias mendelianas, asociación estructurada [30] y análisis de componentes principales [27, 29].
Tercero, covariables como los efectos ambientales y conductuales o el género pueden sesgar los datos. Podemos contabilizarlos incluyéndolos en nuestro modelo estadístico.
Pruebas para la Asociación
Después de realizar los controles de calidad, el análisis estadístico involucrado en GWAS es bastante sencillo, siendo las pruebas más simples la regresión de un solo marcador o una prueba de chi-cuadrado. De hecho, los resultados de asociación que requieren estadísticas arcanas/modelos multimarcadores complejos suelen ser menos confiables.
Primero, asumimos que el efecto de cada SNP es independiente y aditivo para hacer que el análisis sea manejable. Para cada SNP, realizamos una prueba de hipótesis cuya hipótesis nula es que la variación observada en el genotipo en ese SNP entre los sujetos no se correlaciona con la variación observada en el fenotipo entre los sujetos. Debido a que realizamos una prueba para cada SNP, necesitamos lidiar con el problema de múltiples pruebas. Cada prueba tiene alguna probabilidad de dar un resultado falso positivo, y a medida que aumentamos el número de pruebas,
Licencia Commons. Para obtener más información, consulte http://ocw.mit.edu/help/faq-fair-use/.
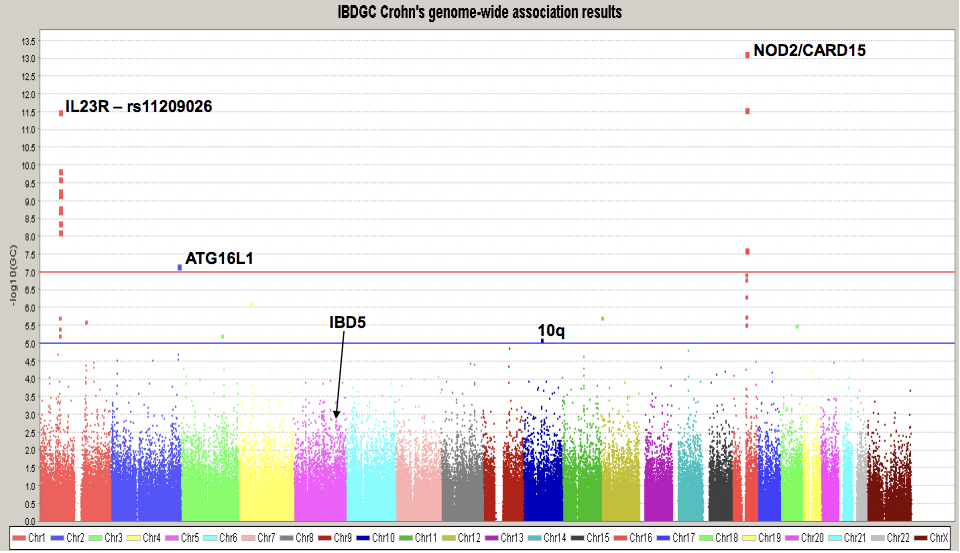
Figura 30.7: Umbrales de significancia de GWAS en la línea azul y líneas rojas para un estudio del IBDGC sobre la enfermedad de Crohn. La línea azul representa un valor p de 5e-8 y la línea roja representa aproximadamente 7.2e-8.
la probabilidad de obtener un falso positivo en cualquiera de ellos aumenta. Esencialmente, con ligamiento, p = 0.001 (.05/ 50 brazos cromosómicos) se consideraría potencialmente significativo, pero GWAS implica realizar pruebas O (10e6) que son en gran medida independientes. Cada estudio tendría cientos de p <0.001 puramente por casualidad estadística, sin relación real con la enfermedad. Existen varios métodos para dar cuenta de múltiples pruebas como la corrección de Bonferroni y medidas como la tasa de falsos descubrimientos [3] y la tasa de descubrimiento irreproducible [22]. Por lo general, la significancia de todo el genoma se establece en p = 5*10e-8 (= .05/1 millón de pruebas), propuestas por primera vez por Risch y Merikangas (1996) []. En 2008, tres grupos [] publicaron estimaciones derivadas empíricamente basadas en mapas densos de todo el genoma de ADN común y estimaron que los números de mapas densos apropiados estaban en el rango de 2.5 a 7.2e-8. Estos se pueden visualizar en la Figura 30.7. Debido a estos diferentes umbrales, es importante observar múltiples estudios para validar asociaciones, ya que incluso con un estricto control de calidad puede haber artefactos que pueden afectar a uno de cada mil o diez mil SNP y evadirse al aviso. Adicionalmente, la significación estricta de todo el genoma generalmente no se excede dramáticamente, si se alcanza, en un solo estudio.
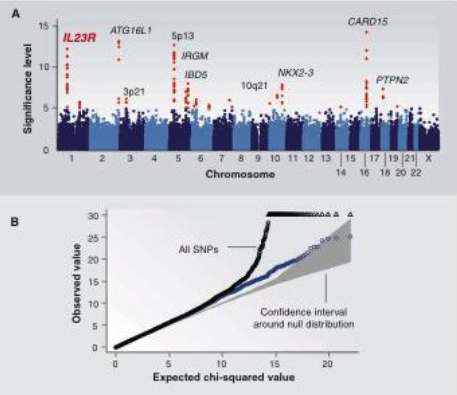
Además de reportar SNP que muestran las asociaciones más fuertes, normalmente también usamos parcelas de Manhattan para mostrar dónde se encuentran estos SNP en el genoma y las parcelas de cuantil-cuantil (Q-Q) para detectar sesgos que no han sido debidamente contabilizados. Una gráfica de Manhattan es una gráfica de dispersión de valores p transformados logarítmicamente contra la posición genómica (concatenando los cromosomas). En la Figura 30.8A, los puntos en rojo son aquellos que cumplen con el umbral de significancia. Se etiquetan con genes candidatos que están cerca. Una gráfica Q-Q es una gráfica de dispersión de los valores p observados transformados logarítmicamente frente a los valores p esperados transformados logar Utilizamos cuantiles uniformes como los valores p esperados: suponiendo que no hay asociación, esperamos que los valores p se distribuyan uniformemente. La desviación de la diagonal sugiere que los valores de p son más significativos de lo esperado. Sin embargo, la desviación temprana y consistente de la diagonal sugiere que demasiados valores p son demasiado significativos, es decir, hay algún sesgo que está confundiendo la prueba. En la Figura 30.8B, la gráfica muestra el estadístico de prueba observado contra el estadístico de prueba esperado (que es equivalente). Considerando todos los marcadores incluye el Complejo Mayor de Histocompatibilidad (MHC), que es la región asociada con la respuesta inmune. Esta región tiene una estructura LD única que confunde el análisis estadístico, como se desprende de la desviación de los puntos negros de la diagonal (el área gris). Tirar el MHC elimina gran parte de este sesgo de los resultados (los puntos azules).
GWAS identifica marcadores que se correlacionan con el rasgo de interés. Sin embargo, cada marcador captura un vecindario de SNP con el que se encuentra en LD, dificultando el problema de identificar la variante causal. Por lo general, el gen candidato para un marcador es el que está más cerca de él. A partir de aquí, tenemos que hacer más estudios para identificar la relevancia de las variantes que identificamos. Sin embargo, esto sigue siendo un problema desafiante por algunas razones:
- Las regiones de interés identificadas por asociación a menudo implican múltiples genes
- Algunas de estas asociaciones no están cerca de ningún segmento codificante de proteínas y no tienen un alelo obviamente funcional como su origen
- Vincular estas regiones con las vías biológicas subyacentes es difícil
Interpretación: ¿Cómo puede el GWAS informar la biología de la enfermedad?
Nuestro objetivo principal es utilizar estas asociaciones encontradas para comprender la biología de la enfermedad de una manera accionable, ya que esto ayudará a guiar las terapias para tratar estas enfermedades. La mayoría de las asociaciones no identifican genes específicos y mutaciones causales, sino que son solo punteros a regiones pequeñas con influencias causales en la enfermedad. Para poder desarrollar y actuar sobre una hipótesis terapéutica, debemos ir mucho más allá, y responder a estas preguntas:
- ¿Qué gen está conectado con la enfermedad?
- ¿Qué proceso biológico está implicado con ello?
- ¿Cuál es el contexto celular en el que ese proceso actúa y es relevante para la enfermedad?
- ¿Cuáles son los alelos funcionales específicos que perturban el proceso y promueven o protegen de enfermedades?
Esto se puede abordar de una de dos maneras: el enfoque de abajo hacia arriba, o el enfoque de arriba hacia abajo.
De abajo hacia arriba
El enfoque de abajo hacia arriba se utiliza para investigar un gen particular que tiene una asociación conocida con una enfermedad, e investigar su importancia biológica dentro de una célula. Kuballa et al. [19] pudieron utilizar este enfoque de abajo hacia arriba para aprender que una variante de riesgo particular asociada a la enfermedad de Crohn conduce a un deterioro de
autofagia de ciertos patógenos. Además, los autores pudieron crear un modelo de ratón de la misma variante de riesgo que se encuentra en humanos. Identificar las implicaciones biológicas de las variantes de riesgo a nivel celular y crear estos modelos es invaluable, ya que los modelos pueden usarse directamente para probar nuevos compuestos potenciales de tratamiento.
De arriba hacia abajo
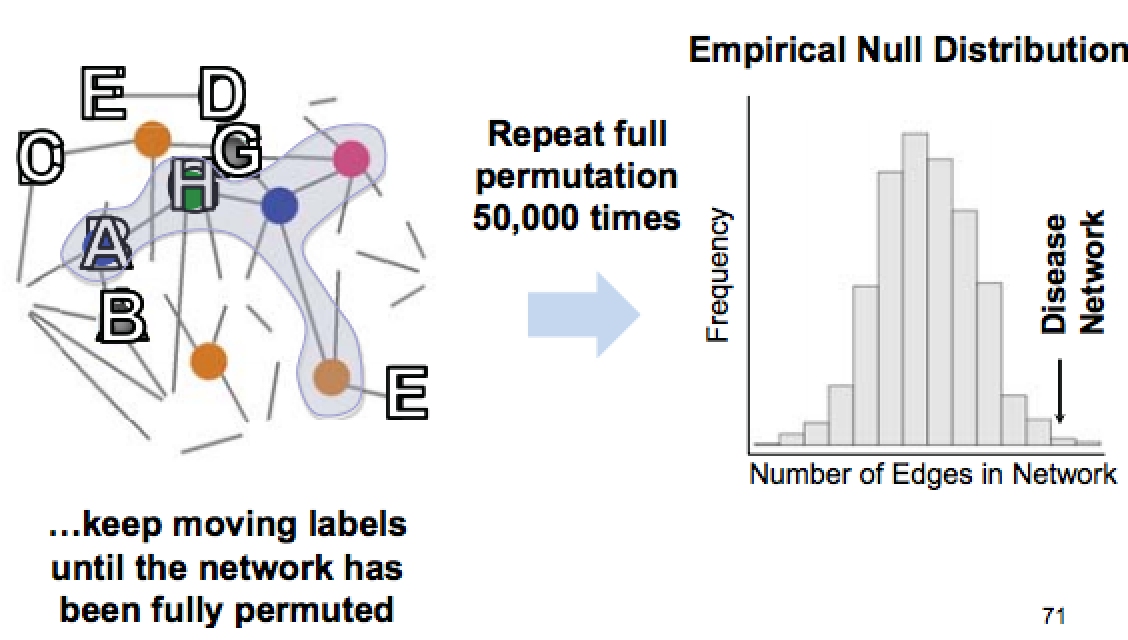
En contraste, el enfoque de arriba hacia abajo implica observar todas las asociaciones conocidas, utilizar el conjunto completo de resultados de GWAS y tratar de vincularlos a procesos/vías biológicas compartidas implicadas en la patogénesis de la enfermedad. Este enfoque se basa en la idea de que muchos de los genes asociados a una enfermedad comparten vías biológicas relevantes. Esto se hace comúnmente tomando redes existentes como redes de interacción proteína-proteína, y superponiendo los genes asociados sobre ellas. Sin embargo, estas redes de enfermedades resultantes pueden no ser significativas debido al sesgo tanto en el descubrimiento de asociaciones como por el sesgo experimental de los datos con los que se están integrando las asociaciones. Esta significación se puede estimar permutando las etiquetas para los nodos en la red muchas veces, y luego calculando cuán raro es el nivel de conectividad para la red de enfermedades dada. Este proceso se ilustra en la Figura 30.9. Como los genes conectados en la red deben ser coexpresados, se ha demostrado que estas redes de enfermedades pueden validarse aún más a partir de perfiles de expresión génica [14].
Comparación con Análisis de Vinculación
Es importante tener en cuenta que GWAS captura más variantes que el análisis de ligamiento. El análisis de ligamiento identifica variantes raras que tienen efectos negativos, y se utilizan estudios de ligamiento cuando se dispone de pedigríes de individuos relacionados con información fenotípica. Pueden identificar alelos raros que están presentes en un número menor de familias, generalmente debido a mutaciones fundadoras y se han utilizado para identificar mutaciones como BRCA1, asociadas con cáncer de mama. Alternativamente, se utilizan estudios de asociación para este propósito y también para encontrar cambios genéticos más comunes que confieren menor influencia en la susceptibilidad, como variantes raras que tienen efectos protectores. El análisis de ligamiento no puede identificar estas variantes porque están anticorrelacionadas con el estado de la enfermedad. Además, el análisis de ligamiento se basa en la suposición de que una sola variante explica la enfermedad, una suposición que no se sostiene para rasgos complejos como la enfermedad. En cambio, necesitamos considerar muchos marcadores para explicar la base genética de estos rasgos.
Si bien la medicina genómica promete nuevos descubrimientos en mecanismos de enfermedades, genes diana, terapéutica y medicina personalizada, quedan varios desafíos, incluyendo que 90+% de los éxitos no son codificantes.
Para solucionar esto, el genoma no codificante ha sido anotado a través de codificación/hoja de ruta y los potenciadores se han vinculado a reguladores y genes diana. Una vez que cada locus GWAS se expande usando el desiquilibrium de ligamiento SNP (LD), se puede usar para reconocer tipos de células relevantes, factores de transcripción de controladores y genes diana. Esto lleva a una vinculación de rasgos con sus tipos relevantes de células y tejidos.
Conclusiones
Hemos aprendido varias lecciones de GWAS. Primero, menos de un tercio de las asociaciones reportadas son variantes codificantes o obviamente funcionales. En segundo lugar, solo algunas fracciones de variantes no codificantes asociadas están significativamente asociadas al nivel de expresión de un gen cercano. En tercer lugar, muchos están asociados a regiones sin gen codificante cercano. Finalmente, la mayoría de las variantes reportadas están asociadas a múltiples enfermedades autoinmunes o inflamatorias. Estas revelaciones indican que todavía hay muchos misterios acechando en el genoma esperando ser descubiertos.