
10.1: Mutaciones - Causas y Significación
- Page ID
- 57166

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
En la célula viva, el ADN sufre frecuentes cambios químicos, especialmente cuando se está replicando (en la fase S del ciclo celular eucariota). La mayoría de estos cambios se reparan rápidamente. Aquellos que no son resultan en una mutación. Así, la mutación es una falla en la reparación del ADN.
Sustituciones de una sola base
Una sola base, digamos una A, se sustituye por otra. Las sustituciones de una sola base también se denominan mutaciones puntuales. (Si una purina [A o G] o pirimidina [C o T] es reemplazada por la otra, la sustitución se denomina transición. Si una purina es reemplazada por una pirimidina o viceversa, la sustitución se denomina transversión).
Mutaciones sin sentido
Con una mutación sin sentido, el nuevo nucleótido altera el codón para producir un aminoácido alterado en el producto proteico.
El reemplazo de A por T en el nucleótido 17 del gen para la cadena beta de la hemoglobina cambia el codón GAG (para ácido glutámico) a GTG (que codifica valina). Así, el 6º aminoácido en la cadena se convierte en valina en lugar de ácido glutámico.
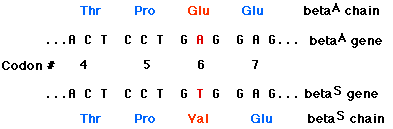
Mutaciones sin sentido
Con una mutación sin sentido, el nuevo nucleótido cambia un codón que especificó un aminoácido a uno de los codones STOP (TAA, TAG o TGA). Por lo tanto, la traducción del ARN mensajero transcrito a partir de este gen mutante se detendrá prematuramente. Cuanto antes en el gen esto ocurra, más truncado es el producto proteico y más probable es que no pueda funcionar.
Aquí hay una muestra de mutaciones que se han encontrado en pacientes con fibrosis quística. Cada una de estas mutaciones ocurre en un enorme gen que codifica una proteína (de 1480 aminoácidos) llamada c ystic f ibrosis t conductancia de transmembrana r egulator (CFTR). La proteína se encarga de transportar iones cloruro y bicarbonato a través de la membrana plasmática. El gen abarca más de 188.000 pares de bases en el cromosoma 7 incrustados en los que hay 27 exones que codifican la proteína. Los números en la columna de mutación representan el número de nucleótidos afectados. Los defectos en la proteína causan los diversos síntomas de la enfermedad. A diferencia de la enfermedad falciforme, entonces, ninguna mutación es responsable de todos los casos de fibrosis quística. Las personas con fibrosis quística heredan dos genes mutantes, pero las mutaciones no necesitan ser las mismas.

En un paciente con fibrosis quística (Paciente B), la sustitución de una T por una C en el nucleótido 1609 convirtió un codón de glutamina (CAG) en un codón STOP (TAG). La proteína producida por este paciente solo tenía los primeros 493 aminoácidos de la cadena normal de 1480 y no pudo funcionar.
Mutaciones silenciosas
La mayoría de los aminoácidos están codificados por varios codones diferentes. Por ejemplo, si la tercera base en el codón TCT para serina se cambia a una cualquiera de las otras tres bases, la serina seguirá siendo codificada. Se dice que tales mutaciones son silenciosas porque no provocan ningún cambio en su producto y no pueden detectarse sin secuenciar el gen (o su ARNm).
Mutaciones en el sitio de empalme
La eliminación de las secuencias de intrones, ya que el pre-ARNm se está procesando para formar ARNm, debe hacerse con gran precisión. Las señales de nucleótidos en los sitios de empalme guían la maquinaria enzimática. Si una mutación altera una de estas señales, entonces el intrón no se elimina y permanece como parte de la molécula de ARN final. La traducción de su secuencia altera la secuencia del producto proteico.
Inserciones y supresiones (indels)
Se pueden agregar pares de bases adicionales (inserciones) o eliminarse (deleciones) del ADN de un gen. El número puede variar de uno a miles. Colectivamente, estas mutaciones se llaman indels.

Los indeles que involucran uno o dos pares de bases (o múltiplos de dos) pueden tener consecuencias devastadoras para el gen porque la traducción del gen se “cambia de marco”. Esta figura muestra cómo desplazando el marco de lectura un nucleótido hacia la derecha, la misma secuencia de nucleótidos codifica una secuencia diferente de aminoácidos. El ARNm se traduce en nuevos grupos de tres nucleótidos y la proteína especificada por estos nuevos codones no tendrá ningún valor. Desplácese hacia arriba para ver otros dos ejemplos (Pacientes C y D).
Los cambios de marco a menudo crean nuevos codones STOP y, por lo tanto, generan mutaciones sin sentido. Quizás eso es tan bien como la proteína probablemente estaría demasiado confusa de todos modos para ser útil a la célula.
Los indeles de tres nucleótidos o múltiplos de tres pueden ser menos graves porque conservan el marco de lectura (ver la figura anterior).
Sin embargo, una serie de trastornos humanos heredados son causados por la inserción de muchas copias del mismo triplete de nucleótidos. La enfermedad de Huntington y el síndrome del X frágil son ejemplos de tales enfermedades de repetición trinucleotídica.
Varios trastornos en humanos son causados por la herencia de genes que han sufrido inserciones de una cadena de 3 o 4 nucleótidos repetidas una y otra vez. Un locus en el cromosoma X humano contiene tal tramo de nucleótidos en el que se repite el triplete CGG (CGGCGGCGGCGG, etc.). El número de CGG puede ser tan pocos como 5 o tantos como 50 sin causar un fenotipo dañino (estos nucleótidos repetidos están en una región no codificante del gen). Incluso 100 repeticiones no suelen causar ningún daño. Sin embargo, estas repeticiones más largas tienen una tendencia a crecer aún más de una generación a la siguiente (hasta 4000 repeticiones).
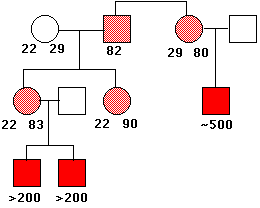
Esto provoca una constricción en el cromosoma X, lo que lo hace bastante frágil. Los machos que heredan tal cromosoma (solo de sus madres, por supuesto) muestran una serie de efectos fenotípicos dañinos, incluido el retraso mental. Las hembras que heredan una X frágil (también de sus madres; los machos con el síndrome rara vez se convierten en padres) solo se ven levemente afectadas.
La imagen anterior muestra el patrón de herencia del síndrome X frágil en una familia. El número de veces que se repite el trinucleótido CGG se da bajo los símbolos. El gen está en el cromosoma X, por lo que las mujeres (círculos) tienen dos copias del mismo; los hombres (cuadrados) tienen solo una. Las personas con un gen que contiene 80—90 repeticiones son normales (rojo claro), pero este gen es inestable, y el número de repeticiones puede aumentar a cientos en su descendencia. Los machos que heredan un gen tan agrandado sufren del síndrome (cuadrados rojos sólidos). (Datos de C. T. Caskey, et al.).
Enfermedades por poliglutamina
En estos trastornos, el trinucleótido repetido es CAG, que agrega una cadena de glutaminas (Gln) a la proteína codificada. Estos han sido implicados en una serie de trastornos del sistema nervioso central, incluyendo
- Enfermedad de Huntington (donde la proteína llamada huntingtina transporta las glutaminas adicionales). La proteína anormal aumenta el nivel de la proteína p53 en las células cerebrales causando su muerte por apoptosis.
- algunos casos de enfermedad de Parkinson donde las glutaminas extra están en la proteína ataxina-2.
Distrofia Muscular
Algunas formas de distrofia muscular que aparecen en adultos son causadas por tri- o tetranucleótidos, por ejemplo (CTG) n y (CCTG) n, repeticiones donde n puede encontrarse con miles. Los enormes transcritos de ARN que resultan interfieren con el empalme alternativo de otros transcritos en el núcleo.
Esclerosis Lateral Amiotrófica (ELA)
La ELA es un trastorno neurodegenerativo que conduce a demencia y debilidad muscular. (La ELA a menudo se llama “enfermedad de Lou Gehrig” por el beisbolista que murió a causa de ella).
La mutación más común en la ELA es una expansión del número de repeticiones del hexanucleótido GGGGCC en un gen en el cromosoma 9 de los dos normales, o al menos menos de tres docenas, a cientos o incluso varios miles. La traducción de las cadenas sentido y antisentido que contienen estas repeticiones (y en los 3 marcos de lectura; no hay codón de inicio ATG) produce polímeros con cadenas largas de gly-ala, gly-pro, gly-arg (de la cadena sentido) así como pro-ala, otro pro-gly y pro-arg de la cadena antisentido. Estas proteínas, especialmente las que contienen arginina (arg) forman agregados que dañan las células cerebrales.
Duplicaciones
Las duplicaciones son una duplicación de una sección del genoma. Durante la meiosis, el cruce entre cromátidas hermanas que están desalineadas puede producir una cromátida con un gen duplicado y la otra (no mostrada) con los dos genes con deleciones. En el caso que se muestra aquí, el cruce desigual creó una segunda copia de un gen necesario para la síntesis de la hormona esteroide aldosterona.

Sin embargo, este nuevo gen porta promotores inapropiados en su extremo 5' (adquiridos del gen 11-beta hidroxilasa) que hacen que se exprese más fuertemente que el gen normal. El gen mutante es dominante: todos los miembros de una familia (a través de cuatro generaciones) que heredaron al menos un cromosoma portador de esta duplicación padecían de hipertensión arterial y eran propensos a la muerte prematura por accidente cerebrovascular.
La duplicación génica también ha sido implicada en varios trastornos neurológicos humanos.
La duplicación génica se ha producido repetidamente durante la evolución de los eucariotas. El análisis del genoma revela muchos genes con secuencias similares en un solo organismo. Presumiblemente estos genes parálogos han surgido por duplicación repetida de un gen ancestral.
Dicha duplicación génica puede ser beneficiosa.
- Con el tiempo, los duplicados pueden adquirir diferentes funciones.
- Las proteínas que codifican pueden asumir diferentes funciones; por ejemplo, si el producto génico original realizó dos funciones diferentes (ver “pleiotropía”), cada gen duplicado ahora puede especializarse en una función y hacer un mejor trabajo en ella que el gen parental.
- Pero aunque no lo hagan, los cambios en las secuencias reguladoras de los genes (promotores y potenciadores) pueden hacer que la misma proteína se exprese en diferentes momentos, en diferentes niveles, y/o en diferentes tejidos.
- Pero aun cuando dos genes parálogos siguen siendo similares en secuencia y función, su existencia proporciona redundancia (“cinturón y tirantes”). Esta puede ser una razón importante por la que noquear genes en levaduras, “ratones knockout”, etc. a menudo tiene un efecto tan leve en el fenotipo. La función del gen noqueado puede ser asumida por un paralog.
- Después de la duplicación génica, pérdida aleatoria, o inactivación, de uno de estos genes en un momento posterior en
- un grupo de descendientes
- diferente a la pérdida en otro grupo
Translocaciones
Las translocaciones son la transferencia de una pieza de un cromosoma a un cromosoma no homólogo. Las translocaciones suelen ser recíprocas; es decir, los dos no homólogos intercambian segmentos.
Las translocaciones pueden alterar el fenotipo de varias maneras:
- la ruptura puede ocurrir dentro de un gen destruyendo su función
- los genes translocados pueden estar bajo la influencia de diferentes promotores y potenciadores para que su expresión sea alterada. La translocación t (8; 14) en el linfoma de Burkitt (figura) es un ejemplo.
- el punto de ruptura puede ocurrir dentro de un gen creando un gen híbrido. Esto puede transcribirse y traducirse en una proteína con un N-terminal de una proteína celular normal acoplada al C-terminal de otra. El cromosoma Filadelfia que se encuentra con tanta frecuencia en las células leucémicas de pacientes con leucemia mielógena crónica (LMC) es el resultado de una translocación que produce un gen compuesto (bcr-abl).
Frecuencia de Mutaciones
Las mutaciones son eventos raros. Esto es sorprendente. Los humanos heredan 3 x 10 9 pares de bases de ADN de cada progenitor. Solo considerando las sustituciones de una sola base, esto significa que cada célula tiene 6 mil millones (6 x 10 9) pares de bases diferentes que pueden ser el objetivo de una sustitución. Las sustituciones de una sola base son más propensas a ocurrir cuando se está copiando el ADN; para eucariotas eso significa durante la fase S del ciclo celular.
Ningún proceso es 100% exacto. Incluso el mecanógrafa más calificado introducirá errores al copiar un manuscrito. Así es con la replicación del ADN. Al igual que un mecanógrafa concienzudo, la célula sí corrige la exactitud de su copia. Pero, aun así, los errores se escapan. Se ha estimado que en humanos y otros mamíferos, los errores no corregidos (= mutaciones) ocurren a razón de aproximadamente 1 de cada 50 millones (5 x 10 7) nucleótidos añadidos a la cadena. (No está mal — Ojalá pudiera escribir con tanta precisión.) Pero con 6 x 10 9 pares de bases en una célula humana, eso significa que cada nueva célula contiene unas 120 nuevas mutaciones.
¿Deberíamos preocuparnos? La evidencia no es clara. Sólo el 1.2% de nuestro ADN codifica los exones de nuestro proteoma, y durante mucho tiempo se pensó que gran parte del resto era ADN “basura”. Las mutaciones en él probablemente serían inofensivas. E incluso en regiones codificantes, la existencia de codones sinónimos podría dar como resultado que el gen alterado (mutado) aún codifique el mismo aminoácido en la proteína. Pero ahora parece que hasta el 80% de nuestro ADN parece participar en regular qué genes se expresan, y con qué fuerza, en cada uno de la multitud de tipos celulares diferenciados en nuestro cuerpo ya que cada uno responde a las señales (nutrientes, hormonas, etc.) que recibe. Entonces, las mutaciones en estas regiones bien podrían tener efectos dañinos, si sutiles.
A medida que se secuencian más genomas de vertebrados, resulta que algunos de estos tramos de ADN que no codifican proteínas, sin embargo, se han conservado notablemente durante la evolución de los vertebrados. Algunas de estas regiones han acumulado incluso menos mutaciones que los genes que codifican proteínas. Esto sugiere que estas secuencias son extremadamente importantes para el bienestar del organismo. Sin embargo, otras regiones del genoma parecen capaces de sostener mutaciones puntuales sin daños detectables.
Los avances recientes han permitido secuenciar las porciones codificantes del genoma de células individuales. Los resultados preliminares indican que cada célula normal en un adulto ha acumulado ~20 mutaciones somáticas, y que su colección de mutaciones difiere de célula a célula. Las células cancerosas acumulan muchas más mutaciones (a menudo en cientos).
¿Cómo podemos medir la frecuencia a la que ocurren las mutaciones que alteran el fenotipo? En los humanos, no es fácil.
- Primero debemos estar seguros de que la mutación es recién surgida. (Algunas poblaciones tienen altas frecuencias de una mutación particular, no porque el gen sea especialmente susceptible, sino porque ha sido transmitido de generación en generación desde un “fundador” temprano.
- Las mutaciones recesivas (la mayoría de ellas son) no se verán salvo en las raras ocasiones en que ambos padres aporten una mutación en el mismo locus a su hijo.
- Esto nos deja con la estimación de frecuencias de mutación para genes que se heredan como
- dominantes autosómicos
- Recesivos ligados al X; es decir, recesivos en el cromosoma X que se expresarán en los machos porque heredan solo un cromosoma X.
Ejemplos
La frecuencia se expresa como la frecuencia de mutaciones que ocurren en ese locus en los gametos
- Dominantes autosómicos
- Retinoblastoma
en el gen RB: alrededor de 8 por millón (8 x 10 -6) - Osteogénesis imperfecta
en uno u otro de los dos genes que codifican colágeno Tipo I: aproximadamente 1 por 100,000 (10 -5) - Tendencia hereditaria a pólipos (y posteriormente cáncer) en el colon.
en un gen supresor de tumores (APC): ~10 -5
- Retinoblastoma
- Recesivos ligados al X
- Hemofilia A
~3 x 10 -5 (el gen del Factor VIII) - Distrofia Muscular de Duchenne (DMD)
>8 x 10 -5 (el gen de la distrofina)
¿Por qué la frecuencia de mutación en el gen de la distrofina debería ser mucho mayor que la mayoría de los demás? Probablemente sea cuestión de tamaño. El gen de la distrofina se extiende sobre 2.4 x 10 6 pares de bases de ADN. ¡Esto es casi 0.1% de todo el genoma humano! Un gen tan enorme ofrece muchas posibilidades de daño.
- Hemofilia A
Medición de la tasa de mutación
La frecuencia con la que se observa una mutación dada en una población (por ejemplo, la mutación que causa la fibrosis quística) proporciona solo una aproximación aproximada de la tasa de mutación, la velocidad a la que ocurren las mutaciones recientes, debido a factores históricos en el trabajo como la selección natural (positivo o negativo), deriva y efecto fundador. Además, la mayoría de los métodos para contar mutaciones requieren que la mutación tenga un efecto visible sobre el fenotipo. Así
- muchas (pero no todas) mutaciones en el ADN no codificante
- mutaciones que producen
- codones sinónimos (codifican el mismo aminoácido)
- o, a veces, nuevos codones que codifican un aminoácido químicamentesimilar
- mutaciones que alteran un gen cuyas funciones son redundantes; es decir, pueden ser compensadas por otros genes
no se verá. Pero ahora estos problemas se han resuelto en gran medida. La historia se cuenta en un reporte de D. R. Denver, et al. en el número 5 de agosto de 2004 de Nature.
El Procedimiento
- Su organismo = C. elegans
- Sus ventajas
- genoma compacto
- hermafrodita — fertiliza sus propios óvulos y cualquier nueva mutación de la línea germinal pronto se perderá o aparecerá en ambos cromosomas homólogos.
- tiempo de generación rápida (4 días)
- Crearon 198 líneas experimentales diferentes de gusanos.
- Los cultivaron en condiciones óptimas para minimizar cualquier efecto de la selección natural.
- Solo se mantuvo una descendencia en cada nueva generación.
- Cada línea se mantuvo durante varios cientos de generaciones.
- Al final de este tiempo, tramos aleatorios de ADN
- derivado de múltiples localizaciones en cada uno de los seis cromosomas de C. elegans y
- totalizando un promedio de ~21 mil pares de bases para cada línea
Resultados
Examinando las secuencias de ADN de sus animales experimentales (¡un total de más de 4 millones de pares de bases!) , y comparándolos con los controles, arrojó un total de 30 mutaciones.
- 17 de ellas fueron inserciones o eliminaciones (“indels')
- 7 en exones, todos menos 2 de los cuales produjeron cambios de marco y un codón PARADA prematuro.
- 10 en intrones o entre genes
- 13 de estas fueron sustituciones de una sola base (mutaciones “puntuales”)
- 3 en exones: uno “silencioso” produciendo un codón sinónimo; dos que cambiaron el aminoácido codificado.
- 10 en intrones o entre genes
Cálculo de tasa de mutación
A partir de estos resultados he agrupado sus datos para calcular una tasa aproximada a la que ocurren mutaciones espontáneas en todo el genoma.
Tasa de Mutación = # de mutaciones observadas [30] ÷ (# de líneas experimentales [198]) x (promedio # de generaciones [339]) x (# promedio de pares de bases secuenciados [~21,000])
produciendo una tasa de 2.1 x 10 -8 mutaciones por par de bases por generación.
El genoma total de C. elegans contiene unos 10 8 pares de bases, por lo que esto nos dice que dos nuevas mutaciones germinales ocurren en algún lugar de cada uno de los dos genomas haploides de C. elegans en cada generación.
Un análisis similar para Drosophila (cuyo genoma es aproximadamente del mismo tamaño que el de C. elegans) mostró una tasa de mutación similar: ~10 -8 mutaciones por par de bases por generación. En cuanto a la planta verde Arabidopsis thaliana, su tasa de mutación espontánea es ligeramente menor: ~7 x 10-9 mutaciones por par de bases por generación.
En la edición del 30 de abril de 2010 de Science, Roach, J. C., et al., reportaron que la tasa para humanos está en el mismo rango: ~1.1 x 10 -8 mutaciones por par de bases en el genoma haploide. Con un genoma diploide de 6 x 10 9 pares de bases, que da resultado a unas 70 nuevas mutaciones en cada niño. Derivaron estos números de la comparación de la secuencia completa del genoma de dos hijos y sus padres.
En la edición del 20 de julio de 2012 de Cell, Wang, J., et al. reportaron los resultados de la secuenciación de 8 espermatozoides individuales de un hombre de 40 años. Encontraron una tasa de mutación que variaba de 2.0 x 10 -8 a 3.8 x 10 -8.
¿Deberíamos preocuparnos por esas tasas de mutación espontánea? Probablemente no demasiado. Con nuestra alta proporción de ADN no codificante, muchas mutaciones ocurrirán en regiones que no tendrán ningún efecto sobre nuestro fenotipo. Evidencia: de un total de 251 mutaciones encontradas en los 8 espermatozoides, solo 3 fueron mutaciones sin sentido alterando un producto génico. Sin embargo, incluso en el ADN no codificante, las mutaciones puntuales pueden afectar la expresión de los genes, por lo que quizás hasta el 10% de las mutaciones puntuales que hereda un niño pueden tener efectos dañinos, si sutiles.
Los machos aportan más mutaciones que las hembras
Si la mayoría de las mutaciones ocurren durante la fase S de la división celular, entonces los machos deberían estar en mayor riesgo. Esto se debe a que solo dos docenas (24) o así ocurren divisiones mitóticas a partir del óvulo fertilizado que inicia el desarrollo embrionario de una niña y la puesta a un lado de sus futuros óvulos (lo cual se hace mucho antes de que ella nazca). Además, los espermatozoides de un hombre de 30 años, en contraste, son descendientes de al menos 400 divisiones mitóticas desde el óvulo fertilizado que lo formó.
Entonces, los padres tienen más probabilidades que las madres de transmitir mutaciones recién formadas a sus hijos. El esperma de un hombre de 25 años podría portar unas 45 nuevas mutaciones. Este número aumenta a un ritmo de aproximadamente 1 por año, por lo que el esperma de un hombre de 40 años puede transmitir unas 60 nuevas mutaciones a sus hijos (alrededor de 20 de estas en regiones codificantes). No importa cuál sea la edad de la madre, transmite solo unas 15 nuevas mutaciones a su descendencia. (Pero las aberraciones cromosómicas, como la aneuploidía, son más propensas a surgir en los óvulos que en los espermatozoides, y la incidencia de estas aumenta con la edad materna). Estos datos explican por qué los hijos de padres ancianos sufren más trastornos genéticos que los de padres jóvenes.
Mutaciones somáticas vs. de línea germinal
La importancia de las mutaciones está profundamente influenciada por la distinción entre línea germinal y soma. Las mutaciones que ocurren en una célula somática, en la médula ósea o en el hígado, por ejemplo, pueden
- dañar la célula
- hacer que la célula sea cancerosa
- matar la celda
Sea cual sea el efecto, el destino final de esa mutación somática es desaparecer cuando muere la célula en la que ocurrió, o su dueño.
Por el contrario, las mutaciones de la línea germinal se encontrarán en cada célula descendiente del cigoto al que contribuyó ese gameto mutante. Si un adulto se produce con éxito, cada una de sus células contendrá la mutación. Entre estos se incluirá la próxima generación de gametos, por lo que si el dueño es capaz de convertirse en padre, esa mutación pasará a otra generación más.