
19.1.1: Taxonomía
- Page ID
- 56967

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
Se han descubierto al menos 1.7 millones de especies de organismos vivos, y la lista crece cada año (especialmente de insectos en la selva tropical). ¿Cómo van a clasificarse? Idealmente, la clasificación debe basarse en la homología; es decir, características compartidas que han sido heredadas de un ancestro común. Cuanto más recientemente dos especies han compartido un ancestro común, más homologías comparten y más similares son estas homologías. Hasta las últimas décadas, el estudio de las homologías se limitaba a las estructuras anatómicas y al patrón de desarrollo embrionario. Sin embargo, desde el nacimiento de la biología molecular, ahora también se pueden estudiar homologías a nivel de proteínas y ADN.
Homología anatómica: un ejemplo
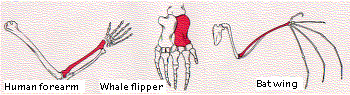
La figura muestra los huesos en las extremidades anteriores de tres mamíferos: humano, ballena y murciélago (¡obviamente no dibujados a la misma escala!). Aunque se utiliza para funciones tan diferentes como lanzar, nadar y volar, el mismo plan estructural básico es evidente en todas ellas. En cada caso, el hueso que se muestra en color es el radio. Las partes del cuerpo se consideran homólogas si tienen
- la misma estructura básica
- la misma relación con otras partes del cuerpo
- se desarrollan de manera similar en el embrión
Parece poco probable que un solo patrón de huesos represente la mejor estructura posible para lograr las funciones a las que se ponen estas extremidades anteriores. Sin embargo, si interpretamos la persistencia del patrón básico como evidencia de herencia de un ancestro común, vemos que las diversas modificaciones son adaptaciones del plan a las necesidades especiales del organismo. Nos dice que la evolución es oportunista, trabajando con materiales que han sido transmitidos por herencia.
Secuencias Proteínas
La secuenciación de proteínas proporciona una herramienta para establecer homologías a partir de las cuales se pueden construir genealogías y dibujar árboles filogenéticos. Aquí hay dos ejemplos.
Hemoglobinas
Un ejemplo de homología molecular.
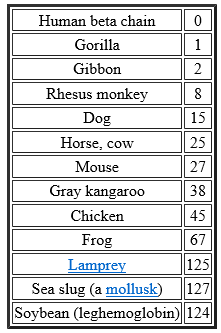
Los números representan el número de diferencias de aminoácidos entre la cadena beta de la hemoglobina humana y las hemoglobinas de las otras especies. En general, el número es inversamente proporcional a la cercanía del parentesco. Todos los valores enumerados son para la cadena beta excepto los tres últimos, en los que no se produce la distinción entre cadenas alfa y beta. La cadena beta humana contiene 146 residuos de aminoácidos, al igual que la mayoría de los demás.
Citocromo c
El citocromo c es parte de la cadena de transporte de electrones por la cual los electrones pasan al oxígeno durante la respiración celular. El citocromo c se encuentra en las mitocondrias de cada eucariota aeróbica: animal, planta y protista. Se han determinado las secuencias de aminoácidos de muchas de estas, y compararlas muestra que están relacionadas. El citocromo c humano contiene 104 aminoácidos, y 37 de estos se han encontrado en posiciones equivalentes en cada citocromo c que se ha secuenciado. Suponemos que cada una de estas moléculas ha descendido de un citocromo precursor en un microbio primitivo que existió hace más de 2 mil millones de años. Es decir, estas moléculas son homólogas.
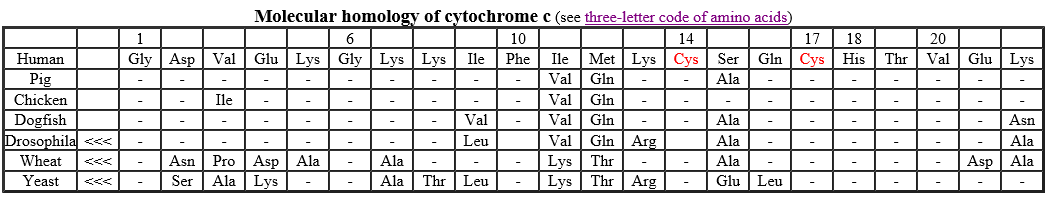
El primer paso para comparar las secuencias del citocromo c es alinearlas para encontrar el número máximo de posiciones que tengan el mismo aminoácido. A veces se introducen brechas para maximizar el número de identidades en la alineación (ninguna fue necesaria en esta tabla). Las brechas corrigen las inserciones y deleciones que ocurrieron durante la evolución de la molécula.
Esta tabla muestra los 22 residuos de aminoácidos N-terminales del citocromo c humano con las secuencias correspondientes de otros seis organismos alineados debajo. Un guión indica que el aminoácido es el mismo que se encuentra en esa posición en la molécula humana. Todos los citocromos vertebrados (los primeros cuatro) comienzan con glicina (Gly). Los citocromos de Drosophila, trigo y levadura tienen varios aminoácidos que preceden a la secuencia aquí mostrada (indicada por <<<). En todos los casos, el grupo hemo del citocromo se une a Cys-14. y Cys-17 (numeración humana). Además de los dos residuos de Cys, Gly-1, Gly-6, Phe-10 e His-18 se encuentran en las posiciones equivalentes en cada citocromo c que se ha secuenciado.
Suponemos que cuantas más identidades haya entre dos moléculas, más recientemente han evolucionado a partir de una molécula ancestral común y, por lo tanto, más cerca está el parentesco de sus dueños. Así, el citocromo c del mono rhesus es idéntico al de los humanos excepto por un aminoácido, mientras que el citocromo c de levadura difiere del de los humanos en 44 posiciones. (No hay diferencias entre el citocromo c de los humanos y el de los chimpancés).
Árboles filogenéticos

Con tal información, se puede reconstruir una historia evolutiva de la molécula y así de sus respectivos dueños. Esto requiere
- usando el código genético para determinar el número mínimo de sustituciones de nucleótidos en el ADN del gen necesario para derivar una proteína de otra
- un potente programa de computadora para buscar los caminos más cortos que unen las moléculas
El resultado es un árbol filogenético. Esta (obra de Walter M. Fitch y Emanuel Margoliash) muestra la relación entre 20 especies de eucariotas. Los números representan el número mínimo de sustituciones de nucleótidos en el gen para el citocromo c necesarias para producir estas 20 proteínas a partir de una serie de genes ancestrales hipotéticos en los diversos puntos de ramificación (nodos).
El árbol corresponde bastante bien a lo que desde hace tiempo hemos creído que son las relaciones evolutivas entre los vertebrados. Pero hay algunas anomalías. Indica, por ejemplo, que los primates (humanos y monos) se separaron antes de la división separando al canguro, un marsupial, de los demás mamíferos placentarios. Esto ciertamente está mal. Pero el análisis de secuencias de otras proteínas puede resolver tales discrepancias.
El citocromo c es una molécula antigua, y ha evolucionado muy lentamente. Incluso después de más de 2 mil millones de años, un tercio de sus aminoácidos permanecen inalterados. Este conservadurismo es de gran ayuda para elaborar las relaciones evolutivas entre criaturas distantemente relacionadas como los peces y los humanos.
Pero, ¿qué pasa con los humanos y los grandes simios? Sus moléculas de citocromo c son idénticas y no nos pueden decir nada sobre las relaciones evolutivas. Sin embargo, algunas proteínas han evolucionado mucho más rápidamente que el citocromo c, y estas pueden usarse para descifrar eventos evolutivos recientes. Durante la coagulación de la sangre, los péptidos cortos se cortan del fibrinógeno convirtiéndolo en fibrina insoluble. Una vez retirados, estos fibrinopéptidos no tienen más función. Han estado prácticamente libres de los rigores de la selección natural y, en consecuencia, han divergido rápidamente durante la evolución. Por lo que aportan datos útiles para clasificar las ramitas de árboles filogenéticos de mamíferos, por ejemplo.
Hibridación ADN-ADN
Como vimos en la comparación del citocromo c humano y canguro, una sola molécula proporciona solo una ventana estrecha para vislumbrar relaciones evolutivas.
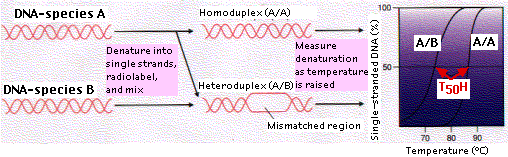
La técnica de hibridación ADN-ADN proporciona una manera de comparar el genoma total de dos especies. Examinemos el procedimiento ya que podría ser utilizado para evaluar la relación evolutiva de la especie B con la especie A:
- El ADN total se extrae de las células de cada especie y se purifica.
- Para cada uno, el ADN se calienta para que se desnaturalice en hebras simples (ssDNA).
- La temperatura se reduce lo suficiente para permitir que las múltiples secuencias cortas de ADN repetitivo se vuelvan a hibridar en ADN bicatenario (ADNbc).
- La mezcla de ADNss (que representa genes individuales) y ADNbc (que representa ADN repetitivo) se pasa sobre una columna empaquetada con hidroxiapatita. El ADNbc se adhiere a la hidroxiapatita; el ADNss no lo hace y fluye directamente a través de ella. El propósito de este paso es poder comparar las porciones codificadoras de información del genoma —en su mayoría genes presentes en una sola copia— sin tener que preocuparse por cantidades variables de ADN repetitivo no informativo.
- El ADNss de la especie A se hace radiactivo.
- El ssDNA radiactivo se permite entonces rehibridar con ssDNA no radiactivo de la misma especie (A) así como —en un tubo separado— con el ssDNA de la especie B.
- Una vez completada la hibridación, las mezclas (A/A) y (A/B) se calientan individualmente en pequeños incrementos (2°—3°C). A cada temperatura más alta, se pasa una alícuota sobre hidroxiapatita. Cualquier cadena radiactiva (A) que se haya separado de los dúplexes de ADN pasa a través de la columna, y la cantidad se mide a partir de su radiactividad.
- Se dibuja una gráfica que muestra el porcentaje de ADNmc a cada temperatura.
- Se determina la temperatura a la que 50% de los dúplexes de ADN (dsDNA) han sido desnaturalizados (T 50 H).
Como muestra la figura, la curva para A/B está a la izquierda de A/A, es decir, dúplexes de A/B separados a una temperatura menor que las de A/A. Las secuencias de A/A son precisamente complementarias por lo que todos los enlaces de hidrógeno entre pares de bases complementarios (A-T, C-G) deben romperse para separar las cadenas. Pero donde las secuencias génicas en B difieren de las de A, no habrá ocurrido ningún emparejamiento de bases y la desnaturalización es más fácil.
Así, la hibridación ADN-ADN proporciona comparaciones genéticas integradas a lo largo de todo el genoma. Su uso ha aclarado varias relaciones taxonómicas desconcertantes. La hibridación ADN-ADN también se puede utilizar para comparar genomas de poblaciones mixtas de organismos. Por ejemplo, cuando todas las bacterias se extraen de 10 g de suelo no contaminado (¡hay alrededor de 10 10 células en él!) , el ADN extraído y purificado de la bacteria y sometido a análisis de hibridación ADN-ADN, las curvas resultantes indican que hay más de un millón de especies diferentes en la muestra de suelo, aunque la población está dominada por solo algunas de estas.
Pintura Cromosómica
Otra forma de comparar genomas completos es unir una etiqueta fluorescente al ADN de cromosomas individuales de una especie (por ejemplo, humano) y exponer los cromosomas de otra especie a él. Las regiones de homología génica se hibridarán tomando la etiqueta fluorescente y los cromosomas “pintados” pueden ser examinados bajo el microscopio.
El método es una modificación de la hibridación fluorescente in situ (FISH) y también se llama Zoo-peces.
La pintura cromosómica ha demostrado, por ejemplo, que grandes secciones del cromosoma 6 humano (que incluye cientos de genes en el complejo mayor de histocompatibilidad (MHC) tienen su contraparte; es decir, genes homólogos, en
- cromosoma 5 del chimpancé
- cromosoma B2 del gato doméstico
- cromosoma 7 del cerdo
- cromosoma 23 de la vaca
Comparación de secuencias de ADN
Las proteínas son la expresión de genes, así que ¿por qué no comparar las secuencias genéticas reales? Hay varias ventajas:
- El ADN es mucho más fácil de secuenciar que la proteína.
- Los genes contienen sitios que son mucho más libres de cambiar durante la evolución que las secuencias de proteínas. Estos incluyen:
- nucleótidos que producen codones sinónimos. Por ejemplo, aunque el aminoácido en la posición 20 en dos proteínas sea el mismo, los codones para ese aminoácido podrían ser diferentes en las dos especies.
- Intrones y secuencias flanqueantes. Estas regiones son relativamente libres de variar sin dañar el producto proteico final. Es decir, estas regiones del genoma están bajo mucha menos presión de la selección natural.
- El ADN es más estable que la proteína en el ambiente. Esto plantea la posibilidad de hacer secuenciación de ADN sobre los restos de organismos extintos. Los restos de Neaderthal de más de 38 mil años han arrojado muestras de ADN que fueron secuenciadas con éxito.
Algunos de los estudios más informativos con secuenciación comparativa de ADN se han realizado con
- Genes de ADNr; es decir, los genes que codifican las moléculas de ARNr (generalmente de la subunidad pequeña (18S en eucariotas; 16S en bacterias) del ribosoma.
- genes en el ADN mitocondrial (ADNmt).
En ambos casos, los genes están presentes en múltiples copias facilitando su aislamiento.
Cladística
Idealmente, un sistema de clasificación debería reflejar las genealogías de los organismos. Darwin se dio cuenta de esto cuando escribió: “nuestras clasificaciones vendrán, en la medida en que así se puedan hacer, genealogías”. Una clasificación basada estrictamente en la regla de que todos los miembros de un grupo deben haber compartido un ancestro común más recientemente que con cualquier especie fuera del grupo se llama cladística.
Este árbol filogenético o cladograma representa las relaciones evolutivas de 4 especies hipotéticas.

- Todos ellos descienden de un antepasado con 5 rasgos (1,2,3,4,5) para ser utilizados en el dibujo del árbol.
- A lo largo del tiempo, ocurrieron 3 eventos de especiación produciendo las ramas.
- Durante este tiempo, varios de los rasgos ancestrales evolucionaron a una forma modificada o derivada; cada uno indicado por un color diferente.
- Los taxonomistas que utilizan métodos cladísticos han creado un vocabulario extraordinario para ayudarlos (no necesariamente a nosotros).
- Los rasgos ancestrales se llaman plesiomórficos (aquí se muestran como números negros).
- Los rasgos derivados se llaman apomórficos (se muestran aquí como números coloreados). Todos los miembros de un clado deben compartir uno o más rasgos apomórficos que no se encuentren en ninguna otra especie.
- Los rasgos derivados compartidos por dos o más especies se denominan sinapomórficos. Aquí las especies A y B comparten el rasgo sinapomórfico designado con un azul 3.
- Los rasgos ancestrales compartidos por dos o más especies se denominan simplesiomórficos. Aquí, el rasgo que se muestra como negro 1 es un rasgo simplesiomórfico retenido por las 4 especies.
- Nótese que al comparar la especie, cuanto más reciente sea el ancestro común, más rasgos apomórficos comparten. Así, las especies C y D comparten 4 de los 5 rasgos pero solo tres (1, 2 y 5) con la especie A y solo dos (1 y 5) con la especie B.
Incluso si reconstruimos una genealogía precisa y dibujamos un árbol filogenético para representarla, los problemas taxonómicos aún pueden permanecer.
- La especie es la única categoría taxonómica que existe en la naturaleza. Todas las categorías superiores (por ejemplo, género, familia y orden) son puramente arbitrarias. Son creados por taxonomistas. Por ejemplo,
- ¿Deben colocarse las especies C y D en un solo género con A y B en otro?
- ¿O están los cuatro lo suficientemente relacionados como para pertenecer a un solo género?
- ¿O los cuatro están tan distantemente relacionados que deberían colocarse en géneros separados?
- Nótese que ninguna de estas opciones (y otras además) viola la regla fundamental de que todos los miembros de cualquier grupo (o "clado “) deben haber tenido un ancestro común más reciente que cualquiera que compartan con especies de otros grupos.

Aquellos taxonomistas que están particularmente impresionados por las diferencias entre especies tienden a aumentar el número de categorías superiores. Aquellos con este sesgo son conocidos con cariño como "divisores”. “Lumpers “, esos taxonomistas que se maravillan de las uniformidades que ven entre las especies, tienden a crear menos categorías superiores. Así, los divisores podrían poner a cada una de las 4 especies en géneros separados, mientras que los terradores las pondrían en un solo género.
- Las clasificaciones basadas estrictamente en la cladística son demasiado complejas para mayor comodidad. En principio, se tiene que crear una categoría separada para todas las ramas derivadas de cada nodo del árbol. El cuadro muestra la clasificación convencional de Homo sapiens (en el orden Primates de la clase Mammalia). Compárelo con el gráfico de arriba del cuadro que muestra una clasificación de solo los primates basada más de cerca en la cladística.
Nombres científicos. El naturalista sueco Carolus Linnaeus -el “padre de la taxonomía ”- creó el sistema para nombrar especies que utilizan los biólogos de todo el mundo. El nombre científico de cada especie consta de dos partes:
- el nombre del género al que está asignado y
- el “epíteto específico” que identifica las especies particulares dentro del género.
Linneo utilizó nombres latinos, pero desde entonces se han descubierto tantas especies que ahora los taxonomistas simplemente acuñan nuevas palabras y lanzan el nombre del género en forma de sustantivo latino y el epíteto específico como adjetivo latino. Por tradición, ambos nombres están impresos en cursiva, y el nombre del género está en mayúscula, pero no el epíteto específico. Tenga en cuenta, también, que los caracteres del alfabeto romano siempre son utilizados incluso por biólogos en países donde se utilizan diferentes caracteres para fines ordinarios.
Aquí hay una descripción de una medusa común tal como aparece en una guía japonesa de vida marina.

Reproducido con permiso de Hoikusha Publishing Co., Ltd., Tokio, Japón
- Una clasificación basada estrictamente en el parentesco evolutivo (cladística) también puede parecer a menudo violar el sentido común. Así, un árbol filogenético que muestra la historia evolutiva que dio origen al salmón (un pez), el pez pulmón y la vaca requiere -según la cladística- que el pez pulmón y la vaca se coloquen en un clado separado del salmón. A pesar de que el pez pulmón es un pez, la vaca ha compartido un ancestro común con él más recientemente que su ancestro común con el salmón. Si bien es tradicional clasificar el pez pulmón y el salmón juntos en la clase Piscis (peces), y asignar la vaca a la clase Mammalia, esto viola la regla de la cladística (por lo que se dice que Piscis es un grupo parafilético). El pez pulmón y la vaca con sus rasgos apomórficos de fosas nasales internas y epiglotis descienden de un ancestro común (flecha roja) que también es el antepasado de todos los vertebrados terrestres (¡incluyéndonos a nosotros mismos!).
Incluso Darwin reconoció que el parentesco por sí solo no siempre era suficiente para una taxonomía sólida por lo que agregó un segundo criterio -grado de similitud- para ser utilizado en la asignación de especies a una categoría taxonómica.
- Deducir la historia evolutiva de los animales es particularmente difícil porque todos los 24 o más filos de los animales aparecieron en poco tiempo antes y durante el Cámbrico y desde entonces han evolucionado a lo largo de líneas separadas. Esto quiere decir que todas las ramas del árbol filogenético son largas y agrupadas tan estrechamente en su base que es difícil determinar sus relaciones.
- Poder de la computadora. Más datos ayudarían, pero a medida que más datos estén disponibles, la capacidad de los programas de computadora para ordenar el árbol más probable se vuelve abrumada.
- Cambio de tasa de evolución. Existe evidencia considerable de que las tasas de mutación no son estables de rama a rama en árboles filogenéticos. Así, una rama basada en moléculas que han evolucionado rápidamente parecería más larga que de otra manera.
- Mutaciones de espalda. Estos enmascaran los cambios que les precedieron y hacen que las ramas parezcan más cortas de lo que deberían ser.
- Transferencia génica entre especies. La reciente disponibilidad de secuencias génicas completas para muchas bacterias ha revelado genes que parecen haber pasado de un grupo a otro en lugar de haber descendido de un ancestro común. La mayoría de estas transferencias de genes “horizontales” son entre dos especies diferentes de bacterias, pero la secuencia génica de Mycobacterium tuberculosis revela 8 genes que parece haber recogido de su huésped humano. Se han producido tantas transferencias de genes horizontales que algunos taxonomistas bacterianos se desesperan de que alguna vez se pueda deducir para ellos un árbol filogenético adecuado.
- Evolución convergente. La evolución en la que dos especies de diferentes genealogías llegan a parecerse entre sí se llama evolución convergente y las estructuras que se asemejan superficialmente (y pueden servir a la misma función) se denominan análogas.

Hay muchos ejemplos de mamíferos marsupiales en Australia que tienen un parecido sorprendente con los mamíferos placentarios de Europa y América del Norte. La marmota o marmota norteamericana y el wombat australiano (foto cortesía del Australian News and Information Bureau), por ejemplo, parecen superficialmente ser parientes cercanos. Pero sus similitudes son análogas, no homólogas, y han surgido como resultado de presiones de selección similares en nichos ecológicos similares. El wombat no tiene placenta, cuida a sus crías en una bolsa como lo hacen otros marsupiales, y debe clasificarse con ellos. De hecho, ¡estamos más estrechamente relacionados con la marmota norteamericana que con el wombat!
En el lenguaje de la cladística, el wombat se coloca en un clado con todos los marsupiales porque comparten la bolsa marsupial (rasgo apomórfico) pero no obstante son mamíferos porque ellos también tienen pelo (un rasgo plesiomórfico).
La evolución convergente también ocurre a nivel de moléculas.
Ejemplos:
- Las vacas y los monos langur sintetizan una lisozima que comparte la misma actividad, pero la comparación de sus secuencias de aminoácidos indica que cada uno ha evolucionado a partir de una molécula ancestral diferente.
- Las vacas y la bacteria Yersinia sintetizan una tirosina fosfatasa con estructuras tridimensionales similares alrededor de su sitio activo y actividad similar. Sin embargo, cada uno ha evolucionado a partir de una molécula ancestral totalmente diferente.
- La bacteria Bacillus subtilis sintetiza una serina proteasa que actúa igual que las sintetizadas por los mamíferos, pero no solo tiene una estructura primaria completamente diferente sino que su estructura tridimensional (terciaria) también es diferente.
- Representantes de cuatro órdenes diferentes de insectos, órdenes que compartieron por última vez un ancestro común hace 300 millones de años, han desarrollado de forma independiente una mutación puntual idéntica en su Na + /K + ATPasa que la protege de la inactivación por los glucósidos cardíacos en las plantas de las que se alimentan. Enlace a una discusión ilustrada sobre cómo esta mutación puede conducir a la coloración aposemática y a la mímica.