
5.2: Buenas Prácticas Clínicas (GCP)
- Page ID
- 53087

El gobierno tiene interés en proteger al público de productos y medicamentos defectuosos. Por lo tanto, las empresas deben demostrar su efectividad y seguridad antes de la distribución masiva. Sin embargo, la única forma en que pueden hacerlo activamente es haciendo que sujetos humanos prueben sus productos.
Si un nuevo producto potencial parece seguro en estudios con animales, entonces se crea un plan para investigar el producto en ensayos clínicos con voluntarios humanos. La compañía presenta su plan a la FDA en un IND. La aplicación IND incluye una descripción del producto, los resultados de las pruebas en animales y los planes para realizar más pruebas. Entonces, la FDA decide si los materiales de la compañía están lo suficientemente completos para que la compañía pueda comenzar a probar el producto en humanos.
¡Explora!
Explore más sobre Buenas Prácticas Clínicas y Ensayos Clínicos:
- http://fda.yorkcast.com/webcast/Play/477af877491747379c36c4ab1c7421b9
- Vea este video sobre los ensayos clínicos Youtu.be/PM1IGF85UOA
Las Buenas Prácticas Clínicas (GCP) se aplican a la realización de ensayos clínicos de seguridad y eficacia de fármacos en sujetos humanos. Las PCG tienen como objetivo proteger los derechos y la seguridad de los sujetos humanos y garantizar la calidad científica de los estudios. Los ensayos clínicos se llevan a cabo por etapas, y cada etapa debe ser exitosa antes de continuar con la siguiente fase. Las Buenas Prácticas Clínicas (GCP) son un conjunto similar de estándares que se aplican a sujetos humanos de ensayos clínicos y experimentos.
Historial regulatorio de las GCP:
- El Código de Núremberg enumera diez principios morales, éticos y legales básicos que describen la investigación médica establecida en respuesta a los juicios del médico de Nuremberg en 1946. Este tribunal inició procesos penales contra médicos por crímenes de lesa humanidad en la Primera Guerra Mundial.
- En 1964, la Asociación Médica Mundial estableció pautas éticas para la investigación biomédica en humanos llamadas Declaración de Helsinki. Estas pautas incluyen códigos de conducta esenciales, incluyendo áreas que involucran consentimiento informado, confidencialidad, revisión de protocolos de investigación, análisis de riesgo versus beneficio, publicación y acceso a datos a la comunidad científica, y la importancia de la salud del sujeto sobre el interés de estudio.
- El Informe Belmont (1979), estableció principios de investigación ética que enfatizan el respeto a las personas, la beneficencia y la justicia, conduciendo a la Regla Común en 1981.
- Para la década de 1980, se hizo evidente que se necesitan poblaciones representativas en los ensayos clínicos, factores que pueden influir en la efectividad y los efectos secundarios de los medicamentos incluyen la edad (niños, pacientes mayores), sexo y etnia.
- En 1989, la FDA emitió directrices pidiendo a los fabricantes determinar si es probable que un medicamento tenga un uso significativo en personas mayores.
- En 1993, la FDA emitió la Guía de Género, que pedía evaluaciones de respuestas a medicamentos en ambos sexos
- En 1998, la FDA requirió que una aplicación de mercadotecnia analice datos sobre seguridad y efectividad por edad, género y raza, conocida como la Regla Demográfica.
- En 2002 se aprobó la Ley de Mejores Productos Farmacéuticos para Niños para mejorar la seguridad y efectividad de los medicamentos para niños.
- En 2003, la FDA recibió clara autoridad bajo la Ley de Equidad en Investigación Pediátrica para exigir a los patrocinadores de medicamentos que realicen investigaciones clínicas sobre aplicaciones pediátricas para nuevos medicamentos.
¡Explora!
Realizar investigaciones someras en internet sobre el Estudio de Sífilis Tuskegee (1932-1972). Resumir por qué este incidente causaría indignación y una disculpa pública por parte de un presidente de Estados Unidos? ¿De qué manera violó esto la Declaración de Helsinki? ¿Cuál fue la respuesta regulatoria? (Es decir, ¿qué ley se aprobó?)
¿Cuáles son las buenas prácticas clínicas?
Si bien no existe una regulación específicamente titulada “Buena Práctica Clínica”, existen varias regulaciones, que rigen la realización de ensayos clínicos.
- Los voluntarios que participen en un estudio clínico deben poder dar su consentimiento informado. Esto significa educar a cada sujeto potencial sobre el tratamiento que debe recibir como parte del estudio así como sobre los riesgos que puedan estar asociados con su participación. La normativa de la FDA titulada “Protecciones de Sujetos Humanos” (21 CFR 50) establece los requisitos para el consentimiento informado.
- Los ensayos clínicos deben ser revisados por un comité independiente del patrocinador del estudio llamado Junta de Revisión Institucional (IRB) (21 CFR 50). En el reglamento se especifica la organización y el personal que integran esta junta, así como los registros e informes que se van a llevar.
- 21 CFR 312 La subparte D describe las responsabilidades de los patrocinadores e investigadores del juicio durante un juicio. Adicionalmente, la “Guía para el Monitoreo de Investigaciones Clínicas” de la FDA explica el monitoreo y la documentación.
¡Pon a prueba tus conocimientos!
Explore MedWatch en el sitio web de la FDA. Luego, vea esta presentación de la FDA en MedWatch:
- ¿Qué es MedWatch? ¿Por qué son importantes?
- Consulta las alertas de seguridad para productos médicos humanos para este año. Discuta una alerta de seguridad que encontró alarmante.
Ética de los Estudios Clínicos
Muchos creen que el 'consentimiento informado' es todo lo que se requiere para satisfacer las preocupaciones éticas de los estudios clínicos. Es mucho más complejo que eso. Además del consentimiento informado, se debe considerar Valor social y clínico, Validez científica, Selección justa de sujetos, Relación riesgo-beneficio favorable, Revisión independiente y Respeto a sujetos potenciales e inscritos. https://clinicalcenter.nih.gov/recruit/ethics.html
El objetivo de la investigación clínica es desarrollar conocimientos generalizables que mejoren la salud humana o aumenten la comprensión de la biología humana. Las personas que participan en la investigación clínica permiten asegurar ese conocimiento. El camino para averiguar si un nuevo medicamento o tratamiento es seguro o efectivo, por ejemplo, es probarlo en pacientes voluntarios. Sin embargo, al poner a algunas personas en riesgo de sufrir daños por el bien de otras, la investigación clínica tiene el potencial de explotar a pacientes voluntarios. El propósito de las pautas éticas es tanto proteger a los pacientes voluntarios como preservar la integridad de la ciencia.
Las pautas éticas vigentes hoy en día fueron principalmente una respuesta a abusos pasados, el más notorio de los cuales en América fue un experimento en Tuskegee, Alabama, en el que se retuvo el tratamiento a 400 hombres afroamericanos con sífilis para que los científicos pudieran estudiar el curso de la enfermedad. En el siglo XX se desarrollaron diversos lineamientos éticos en respuesta a dichos estudios.
El informe Belmont
Existen muchas pautas además de reglas y regulaciones que rigen la ética de los estudios clínicos. Algunos de los más influyentes incluyen El Código de Nuremberg (1947), Declaration of Helsinki (2000), Belmont Report (1979), CIOMS (2002) y US Common Rule (1991). Lea el Informe Belmont aquí: https://www.hhs.gov/ohrp/regulations-and-policy/belmont-report/index.html
Consentimiento Informado
Cualquier paciente que participe en un estudio clínico deberá hacerlo bajo consentimiento informado. Algunas excepciones a esta regla incluyen operaciones militares o emergencias de salud pública. El consentimiento informado a la FDA no solo incluye la autorización del paciente sino un intercambio de información entre el sujeto y el individuo que obtiene esta aprobación. El sujeto debe tener suficiente información sobre el estudio para tomar una decisión informada sobre su participación en el estudio. El consentimiento informado se describe en el Formulario de Consentimiento Informado (ICF), permite al sujeto tiempo para reflexionar y tiene la información disponible para hacerlo, y por lo tanto, el ICF se somete a la FDA para su revisión.
Junta de Revisión Institucional (IRB)
Una empresa también debe obtener la aprobación de una Junta de Revisión Institucional (IRB) para realizar pruebas en humanos. El IRB es un grupo responsable de proteger los derechos, la seguridad y el bienestar de los sujetos humanos. Por lo general, está compuesto por un mínimo de cinco miembros con diversidad de género; al menos un miembro científico y un miembro no científico. Los estándares generales del IRB están cubiertos y descritos en 21 CFR Parte 56. La FDA tiene una lista completa de regulaciones que rigen Estudios Clínicos (ClinicalStudies.gov). Aquí también se encuentran documentos de orientación internacional de GCP, en los que la FDA ha colaborado, y enlaces a otros sitios relevantes para la realización de ensayos clínicos, tanto a nivel nacional como internacional.
Monitoreo de Bioinvestigación
Los objetivos generales del programa de monitoreo de bioinvestigación (BIMO) de la FDA son proteger los derechos, la seguridad y el bienestar de los sujetos involucrados en ensayos clínicos regulados por la FDA; determinar la precisión y confiabilidad de los datos de ensayos clínicos presentados a la FDA; y evaluar el cumplimiento con las regulaciones de la FDA que rigen la realización de ensayos clínicos, incluidos aquellos para el consentimiento informado y la revisión ética. El programa BIMO realiza inspecciones in situ de estudios clínicos y no clínicos realizados para apoyar las aplicaciones/envíos de investigación y mercadotecnia a la agencia.
Declaración de misión del Consultorio de Buena Práctica Clínica
La Oficina de Buena Práctica Clínica es el punto focal dentro de los temas de la FDA para la Buena Práctica Clínica (GCP) y la Protección del Sujeto Humano (HSP) que surgen en ensayos de investigación en humanos regulados por la FDA.
Inicio del estudio clínico
Un patrocinador necesita lo siguiente para iniciar un estudio clínico:
- IRB
- Documentación de las credenciales del investigador clínico
- Declaración de la situación financiera (otorgante-patrocinador)
- Declaraciones de aseguramiento GCP
- Verificación de protocolo de estudio
Informes de estudios clínicos
El investigador debe proporcionar informes de progreso del estudio clínico a intervalos específicos durante el estudio.
Diseño de Estudio Clínico
Un estudio clínico es cualquier estudio de investigación que involucra a uno o más sujetos humanos que prueban nuevos fármacos experimentales, dispositivos o biológicos (o control). Es tarea del investigador diseñar el protocolo de estudio clínico. Hay dos tipos principales de ensayos clínicos; estudios clínicos (intervencionistas) y estudios observacionales. Para ciertos dispositivos médicos, los estudios de precisión también pueden ser apropiados. Dentro del tipo de estudio clínico, existen varios subtipos, que pueden incluir controles de placebo, estudios doble ciego y controles de aleatorización.
¡Pon a prueba tus conocimientos!
Beth es una mujer con discapacidad mental posmenopáusica de 46 años con LCIS, es decir, está predispuesta a desarrollar cáncer de mama más adelante en la vida. Sus cuidadores con poder notarial para decisiones de atención médica, la llevan a la clínica para su inscripción en un ensayo clínico, que es un ensayo aleatorizado de tamoxifeno v. raloxifeno para la prevención del cáncer de mama en mujeres de alto riesgo. Cumple con todos los requisitos de ingreso pero no puede consentir debido a su discapacidad mental. El IRB está considerando, ¿es ético permitir que el poder notarial de Beth la inscriba en este estudio? *Recuerda, aquí nos estamos enfocando en la ética, no en la ley*.
- ¿Qué es un IRB? ¿Cuál es su función?
- Usando lo que has aprendido en este capítulo sobre GCP y los recursos éticos a continuación, argumenta a favor de OR en contra del fallo del IRB para otorgarle permiso al poder notarial de Beth para inscribirla en el estudio.
Ensayos Clínicos
Durante un ensayo clínico, los participantes pueden recibir una intervención particular, un nuevo fármaco en investigación, dispositivo o biológico, o incluso tratamiento psicológico, por ejemplo, dieta o dejar de fumar. Estas intervenciones pueden ser una comparación de un fármaco actual con un nuevo fármaco en investigación, un placebo sin ingrediente activo con un fármaco existente, por citar algunos ejemplos. El ensayo también puede ser aleatorizado, placebocontrol y cegado para reducir el sesgo del estudio.
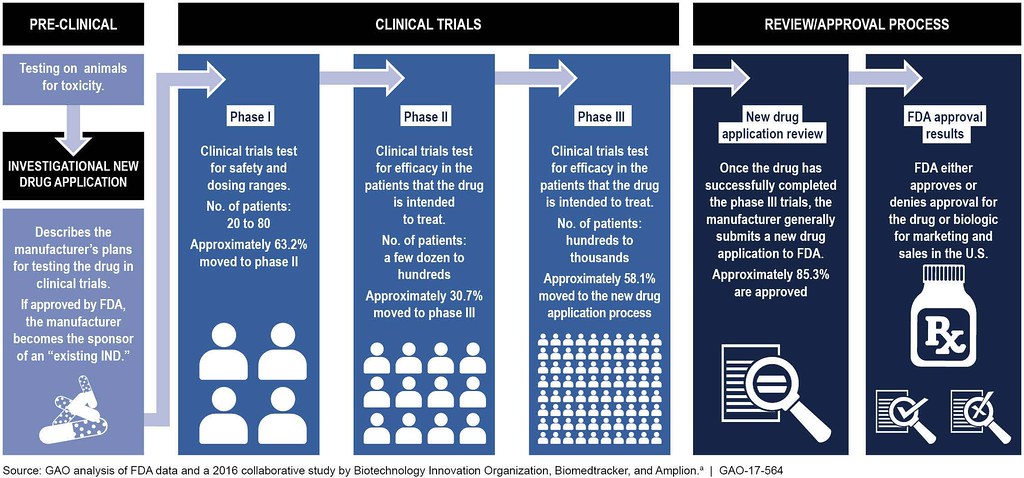
Las cuatro fases de los ensayos clínicos
- En Estudios Preclínicos, el medicamento se prueba en animales para determinar su seguridad y eficacia.
- Los ensayos clínicos de fase I prueban principalmente la seguridad del fármaco propuesto en humanos sanos. Durante los ensayos de Fase I, el medicamento se administra a 20-80 voluntarios sanos que reportarán cualquier efecto secundario inesperado y ayudarán a establecer los niveles de dosificación que se pueden tolerar. Además de evaluar la seguridad del fármaco, se determinan sus propiedades metabólicas y farmacológicas en humanos sanos. Si un medicamento cumple con los requisitos de seguridad en esta fase y parece tener el impacto deseado del tratamiento, entonces ingresa a los ensayos clínicos de Fase II.
- Los ensayos de fase II se realizan en un pequeño número de pacientes para determinar la eficacia del medicamento. Entre 100 y 300 pacientes que el medicamento se pretende tratar reciben diversas dosis. Los participantes del ensayo clínico son monitoreados cuidadosamente para detectar los efectos secundarios, así como las consecuencias del tratamiento farmacológico. Si no hay efectos secundarios perjudiciales, y el medicamento tiene un efecto positivo, pasa a la fase III.
- Los ensayos de fase III involucran entre 1,000 y 3,000 pacientes en estudios doble ciego generalmente realizados en varios sitios hospitalarios. Para la mayoría de los medicamentos nuevos, estas pruebas durarán tres o más años para establecer los beneficios del medicamento, la dosis recomendada y la seguridad a largo plazo. Se recolectan datos adicionales sobre interacciones medicamento-drogas y riesgos versus beneficios.
- Los ensayos de fase IV se realizan después de que el medicamento haya sido aprobado por la FDA. Esta vigilancia poscomercialización ayuda a recopilar información adicional sobre la seguridad y eficacia de los medicamentos por parte de la población en general.
Ensayos clínicos de dispositivos médicos
No todos los dispositivos médicos se someten a pruebas de ensayo clínico. Los dispositivos de riesgo mínimo como los vendajes (Clase I) no requieren ensayos clínicos, donde los dispositivos Clase II de riesgo intermedio pueden depender del dispositivo. Los dispositivos de clase III tienen un riesgo sustantivo y por lo tanto se someten a ensayos clínicos. Otra diferencia entre los ensayos clínicos con dispositivos médicos es lo que se prueba. Para los medicamentos, se prueba una dosis; sin embargo, en los dispositivos lo es el prototipo. Dado que existe una variedad de tipos de dispositivos médicos, lo exploraremos más a fondo en un capítulo posterior centrado en los dispositivos médicos.
El NIH tiene un registro de acceso público de todos los ensayos clínicos actualmente en curso e incluye los resultados, apropiadamente denominados ClinicalTrials.gov.
¡Explora!
Ve a ClinicalTrials.gov y “busca un estudio” de algo que te interese. Por ejemplo, si has estado siguiendo el último brote del virus del Ébola o Zika, tal vez te preguntes sobre el estado de cualquier estudio de vacunas actual. Escribe un resumen de 5 frases del estudio que seleccionaste.