
7.2: Clasificación sin aprendizaje
- Page ID
- 149995

Vemos que el trazado de datos multivariados siempre tiene dos problemas: o hay demasiados elementos (por ejemplo, en coordenadas paralelas) que son difíciles de entender, o hay necesidad de alguna operación de agrupación (por ejemplo, mediana o rango) que resultará en la pérdida de información. Lo que será realmente útil es procesar primero los datos de manera segura, por ejemplo, para reducir las dimensiones —de muchas a 2 o 3. Estas técnicas se describen en esta sección.
Aparte de (a) reducción de dimensionalidad (búsqueda de proyección), los siguientes métodos ayudan a (b) encontrar grupos (clusters) en los datos, (c) descubrir factores ocultos (variables latentes) y comprender la importancia de las variables (selección de características\(^{[1]}\)), (d) reconocer objetos (por ejemplo, formas complicadas) dentro de los datos, típicamente usando densidades e hiato (huecos) en el espacio multidimensional, y (e) señales de desmezcla.
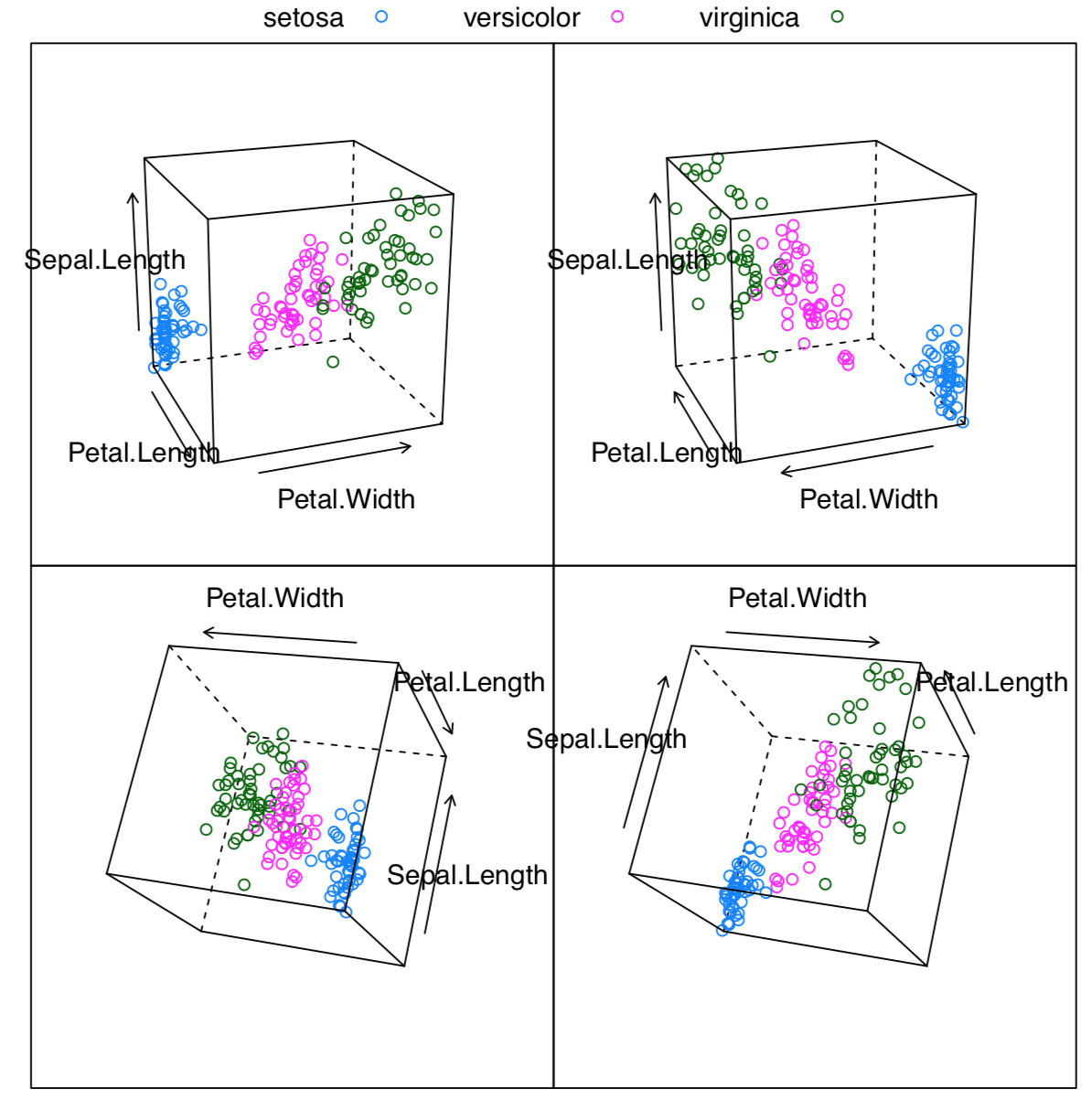
Clasificación con datos primarios
Primario es lo que viene directamente de la observación, y aún no procesa de ninguna manera (para hacer datos secundarios).
Sombras de hiper nubes: PCA
RGL (ver arriba) permite encontrar la mejor proyección manualmente, con un ratón. Sin embargo, es posible hacer programáticamente, con análisis de componentes principales, PCA. Pertenece a la familia de métodos no supervisados, métodos de clasificación sin aprendizaje, u ordenación.
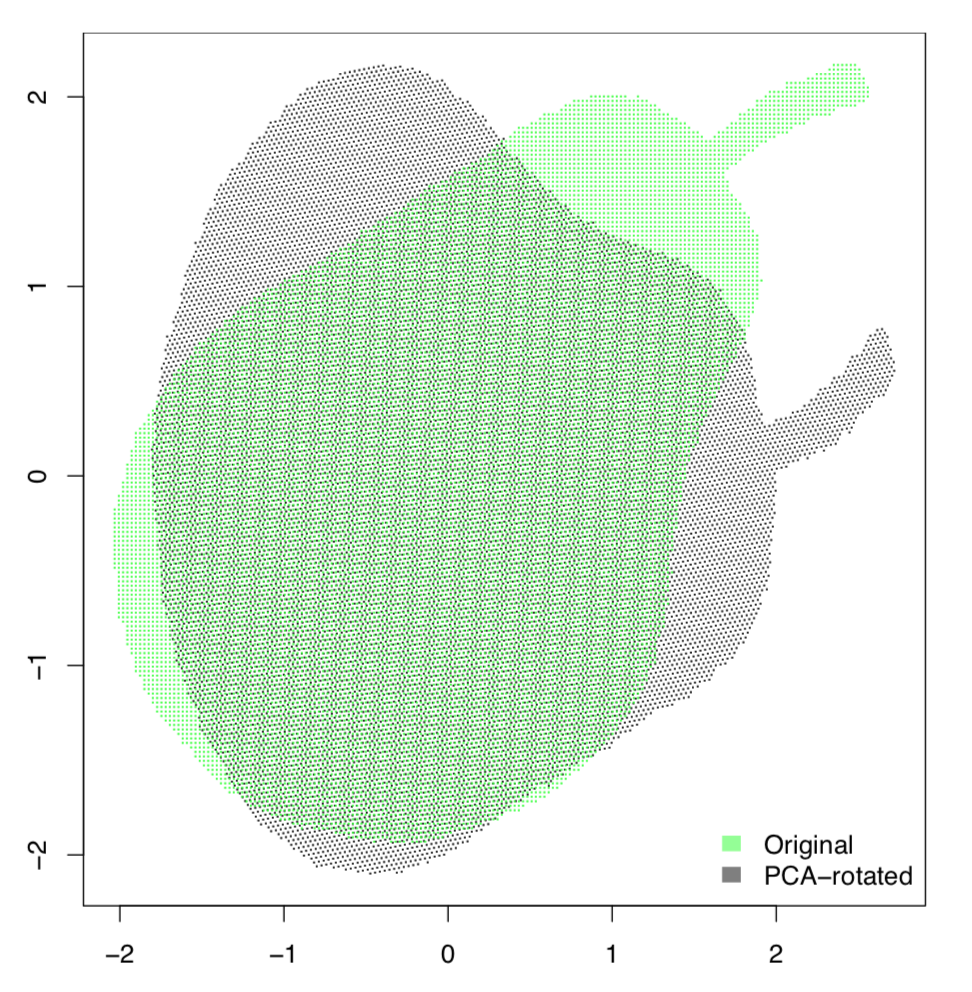
PCA trata los datos como puntos en el espacio virtual multidimensional donde cada dimensión es el carácter único. Estos puntos conforman en conjunto la nube multidimensional. El objetivo del análisis es encontrar una línea que cruce esta nube a lo largo de su parte más alargada, como pera en el palo (Figura\(\PageIndex{2}\)). Este es el primer componente principal. La segunda línea es perpendicular a la primera y nuevamente abarca la segunda parte más alargada de la nube. Estas dos líneas conforman el plano sobre el que se proyecta cada punto.
Demostrémoslo prácticamente. Cargaremos la imagen de pera bidimensional (de ahí solo dos componentes principales) en blanco y negro y veremos qué hace PCA con ella:
Código\(\PageIndex{1}\) (R):
PCA se relaciona con una tarea de encontrar a la “persona más promedio”. La simple combinación de promedios no funcionará, lo cual está bien explicado en el libro “El fin del promedio” de Todd Rose. Sin embargo, generalmente es posible encontrar en el hiperespacio la configuración de parámetros que se adaptarán a la mayoría de las personas, y para esto es para lo que sirve PCA.
Después del procedimiento PCA, todas las columnas (caracteres) se transforman en componentes, y el componente más informativo es el primero, el siguiente es el segundo, luego el tercero etc. El número de componentes es el mismo que el número de caracteres iniciales pero los primeros dos o tres suelen incluir toda la información necesaria. Es por ello que es posible utilizarlos para la visualización 2D de datos multidimensionales. Hay muchas similitudes entre el ACP y el análisis factorial (que está fuera del alcance de este libro).
Al principio, usaremos un ejemplo del repositorio abierto presentando mediciones de cuatro poblaciones diferentes de juncias:
Código\(\PageIndex{2}\) (R):
(Escala de función () estandariza todas las variables.)
La siguiente gráfica (Figura\(\PageIndex{4}\)) es una gráfica de pantalla técnica que muestra la importancia relativa de cada componente:
Código\(\PageIndex{3}\) (R):
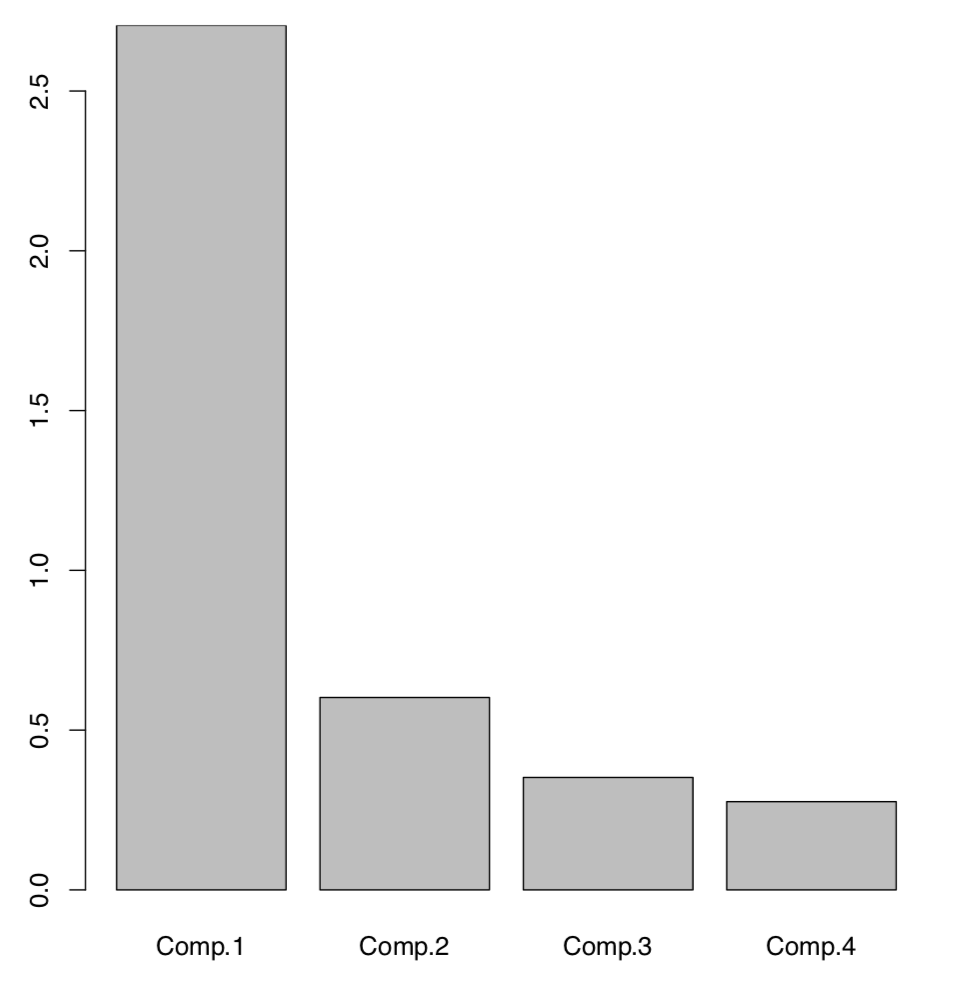
Aquí es fácil ver que entre cuatro componentes (mismo número que los caracteres iniciales), dos primeros tienen las mayores importancias. Hay una manera de tener lo mismo sin trazar:
Código\(\PageIndex{4}\) (R):
Los dos primeros componentes juntos explican alrededor del 84% por ciento de la varianza total.
La visualización de PCA generalmente se realiza utilizando puntuaciones del modelo PCA (Figura\(\PageIndex{5}\)):
Código\(\PageIndex{5}\) (R):
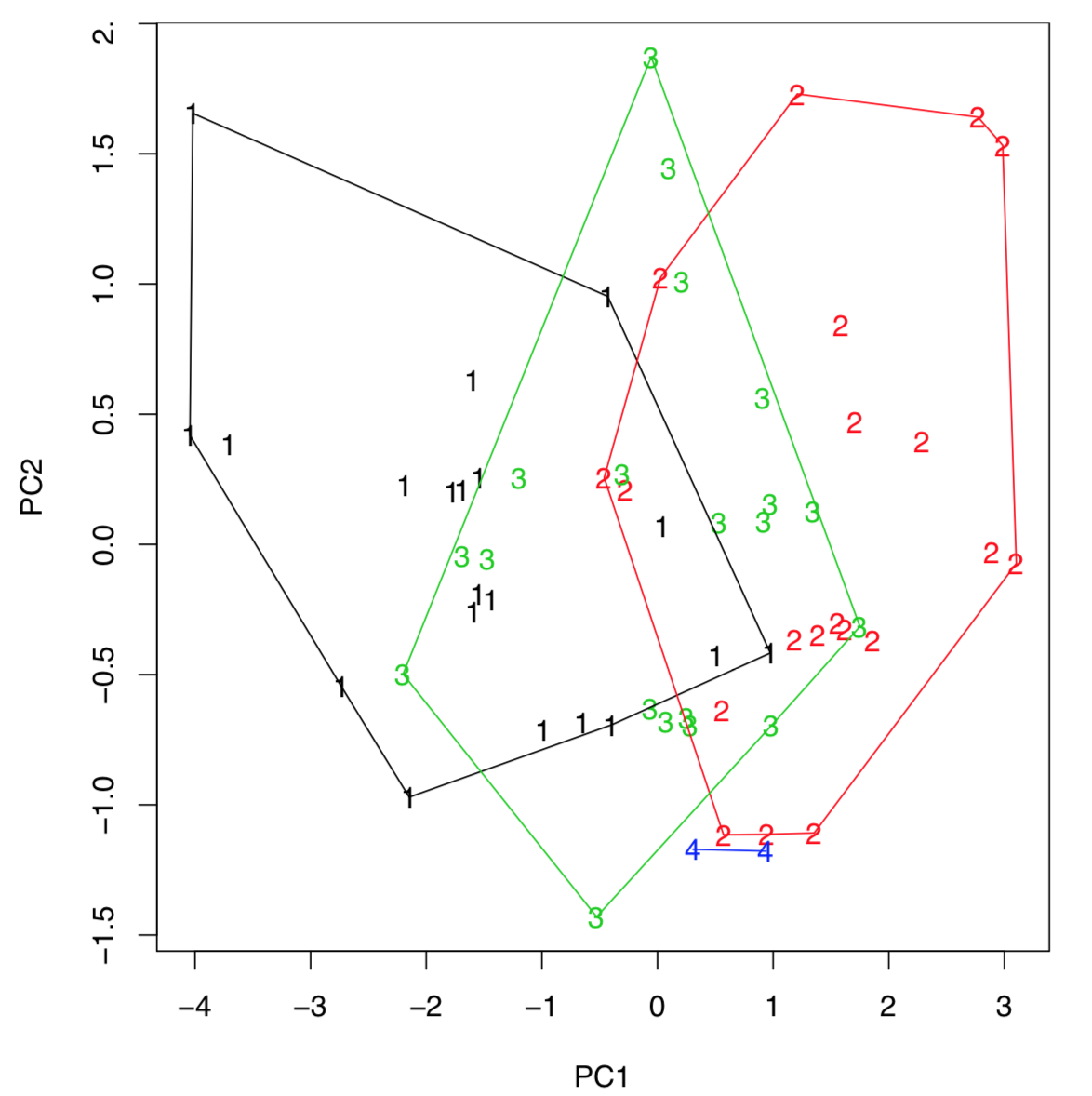
(El último comando dibuja cascos que ayudan a concluir que las primeras juncias de la tercera población son intermedias entre la primera y la segunda, podrían ser incluso híbridos. Si hay tres, no dos, componentes que son los más importantes, entonces cualquiera de las gráficas 3D como scatterplot3d () explicadas anteriormente, ayudará a visualizarlos.)
Es tentador medir la intersección entre cascos. Esto es posible con la función Overlap (), que a su vez carga el paquete PBSMapping:
Código\(\PageIndex{6}\) (R):
En ocasiones, los resultados de PCA son útiles para presentar como biplot (Figura\(\PageIndex{6}\)):
Código\(\PageIndex{7}\) (R):
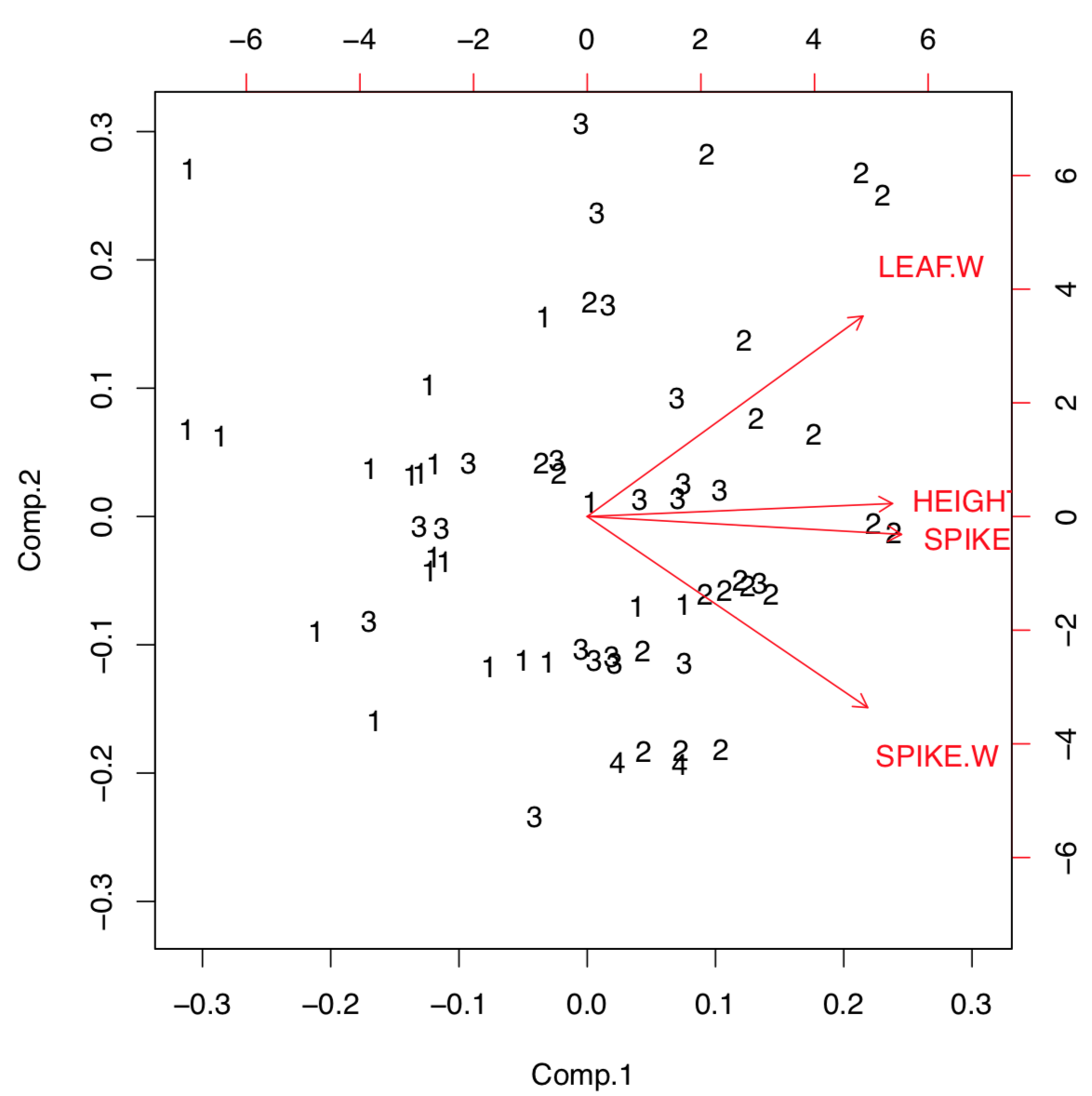
Biplot ayuda a comprender visualmente qué tan grande es la carga de cada carácter inicial en los dos primeros componentes. Por ejemplo, los caracteres de altura y longitud de espiga (pero ancho de espiga) tienen una mayor carga en el primer componente que distingue más a las poblaciones. Cargas de funciones () permite ver esta información en forma numérica:
Código\(\PageIndex{8}\) (R):
R tiene dos variantes de cálculo de PCA, primero (ya discutido) con princomp () y segundo con prcomp (). La diferencia radica en la forma en que exactamente se calculan los componentes. La primera forma es tradicional, pero se recomienda la segunda:
Código\(\PageIndex{9}\) (R):
El ejemplo anterior muestra algunas diferencias entre dos métodos de PCA. Primero, prcomp () acepta convenientemente la opción de báscula. Segundo, las cargas se toman del elemento de rotación. Tercero, las puntuaciones están en el elemento con x nombre. Por favor, ejecute el código usted mismo para ver cómo agregar elipses de confianza del 95% a la gráfica de ordenación 2D. Se puede ver que Iris setosa (letra “s” en la parcela) es seriamente divergente de otras dos especies, Iris versicolor (“v”) e Iris virginica (“a”).
Los paquetes ade4 y veganos ofrecen muchas variantes de PCA (Figura\(\PageIndex{7}\)):
Código\(\PageIndex{10}\) (R):
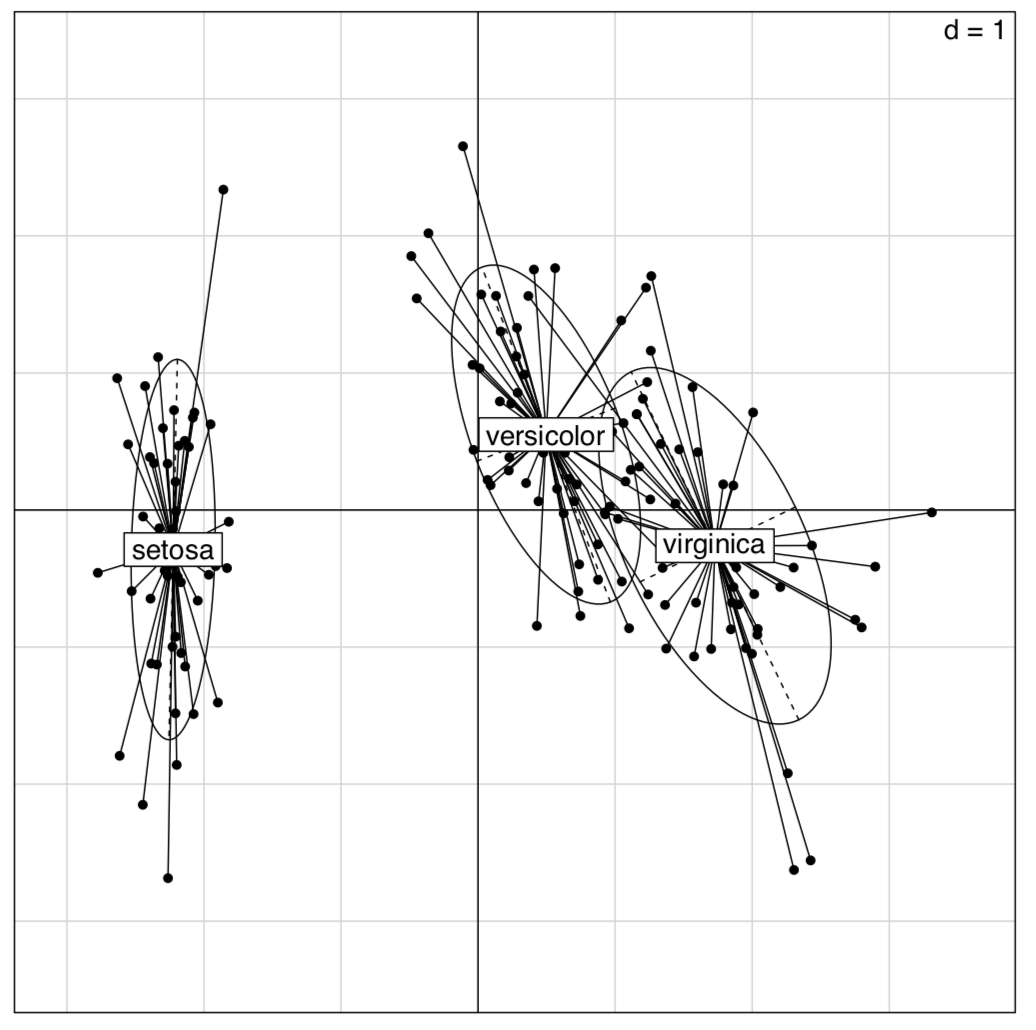
(La gráfica es similar a la mostrada en la Figura\(\PageIndex{5}\); sin embargo, las diferencias entre grupos son aquí más claras.)
Además, esto es posible utilizar el enfoque inferencial para el PCA:
Código\(\PageIndex{11}\) (R):
La aleatorización de Montecarlo permite comprender numéricamente qué tan bien se separan las especies de Iris con este PCA. El alto valor de Observación (72.2% que es mayor al 50%) es el signo de diferencias confiables.
Existen otras variantes de pruebas de permutación para PCA, por ejemplo, con anosim () del paquete vegano.
Tenga en cuenta que el análisis de componentes principales es en general una técnica lineal similar al análisis de correlaciones, y puede fallar en algunos casos complicados.
Solitario de datos: SOM
Existen varias otras técnicas que permiten la clasificación no supervisada de los datos primarios. Los mapas autoorganizados (SOM) son una técnica algo similar a romper la baraja de cartas en varios montones:
Código\(\PageIndex{12}\) (R):
La gráfica resultante (Figura\(\PageIndex{8}\)) contiene representación gráfica de valores de caracteres, junto con la colocación de puntos de datos reales.
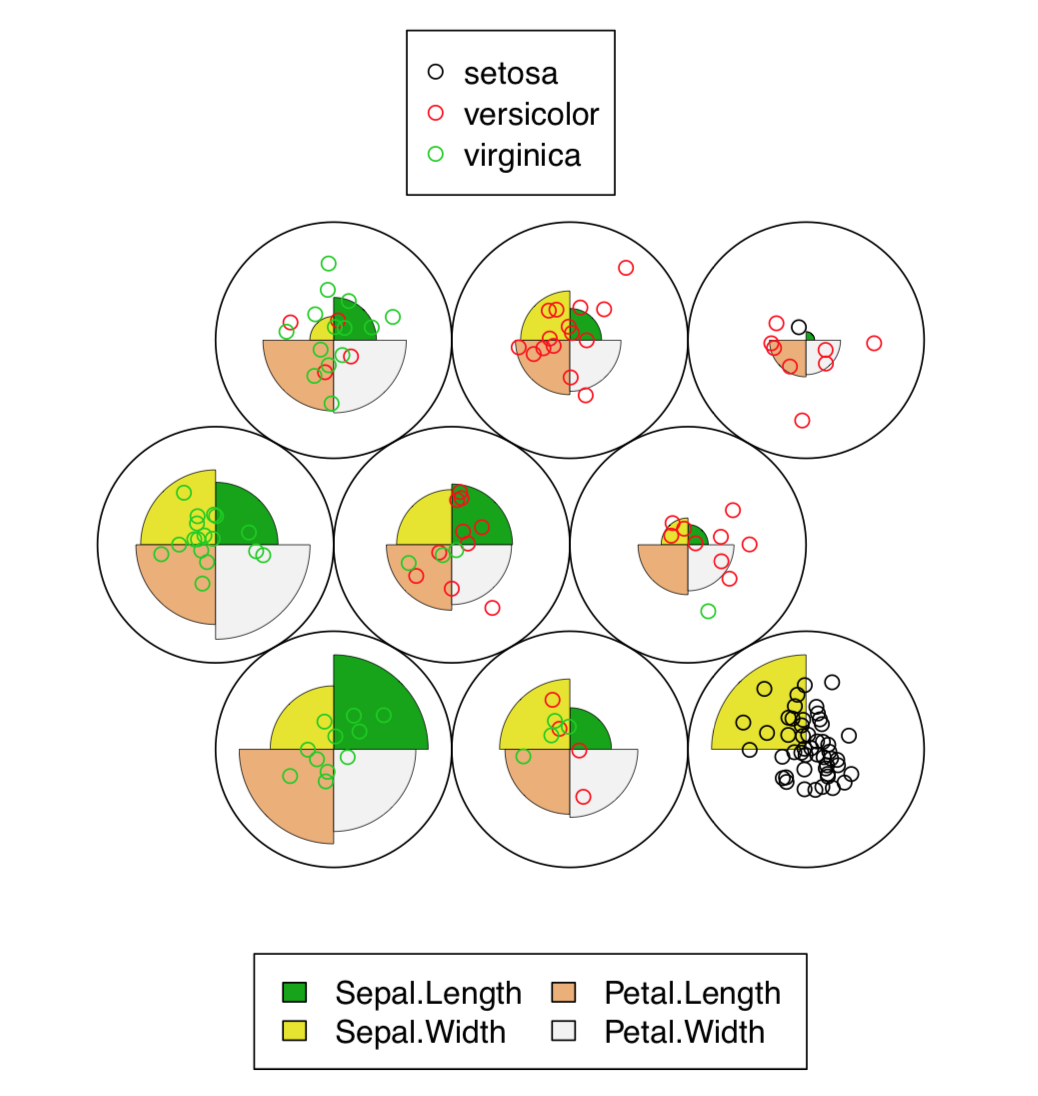
De hecho, SOM es la red neuronal que no aprende. El algoritmo más avanzado de Gas Neural en Crecimiento (GNG) utiliza ideas similares a SOM.
Densidad de datos: T-SNE
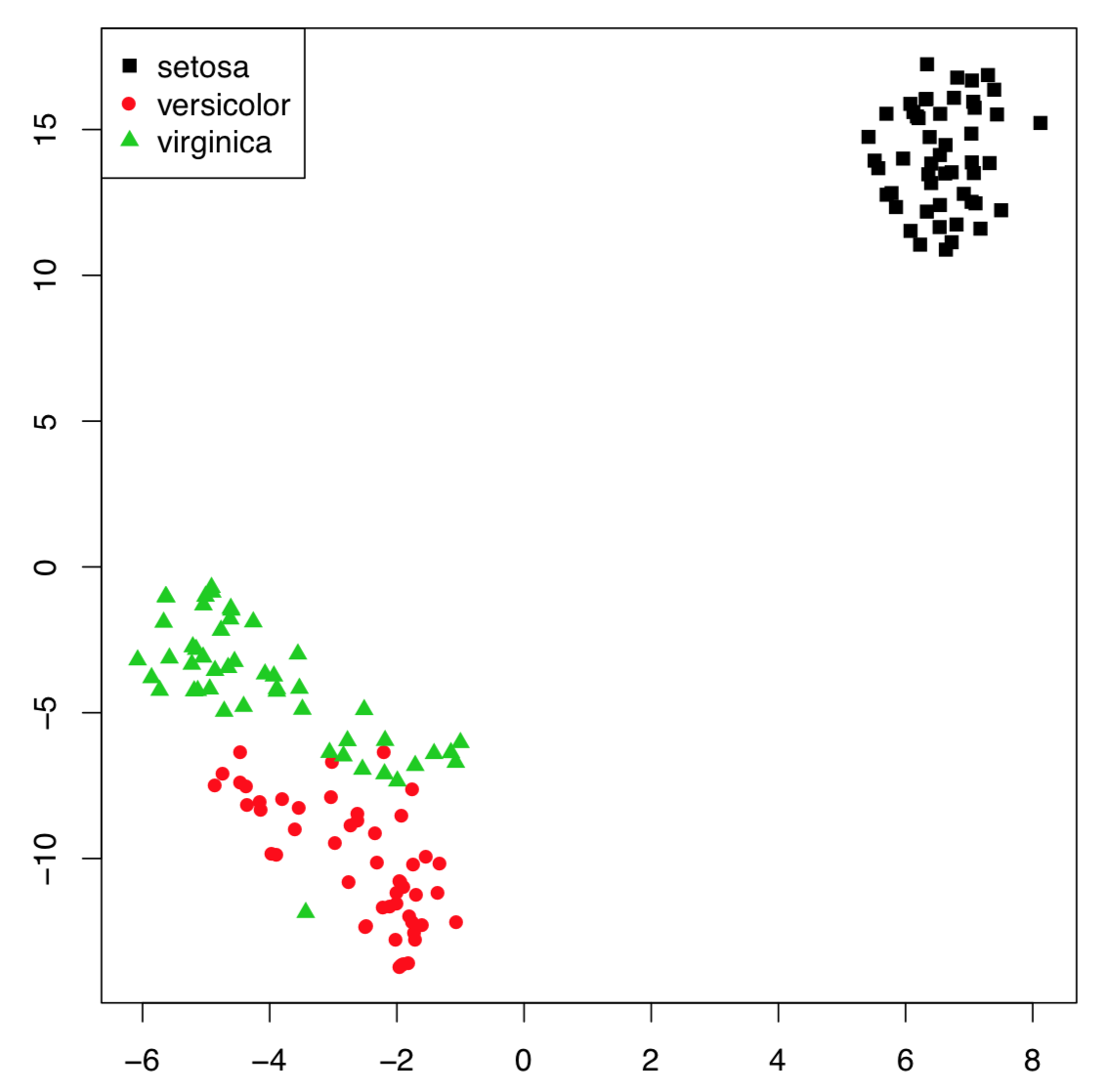
Con el gran número de muestras, el algoritmo T-SNE (nombre significa “Embedding Estocástico Vecino Distribuido en T”) funciona mejor que el PCA clásico. t-SNE se usa con frecuencia para el reconocimiento de formas. Es bastante fácil emplearlo en R (Figura\(\PageIndex{9}\)):
Código\(\PageIndex{13}\) (R):
Clasificación con correspondencia
El análisis de correspondencia es la familia de técnicas similares al PCA, pero aplicables a datos categóricos (primarios o en tablas de contingencia). La variante simple del análisis de correspondencia se implementa en corresp () a partir del paquete MASS (Figura\(\PageIndex{10}\)) que trabaja con tablas de contingencia:
Código\(\PageIndex{14}\) (R):
(Convertimos aquí objeto “tabla” HE en el marco de datos. XPD=true se utilizó para permitir que el texto saliera del cuadro de trazado.)
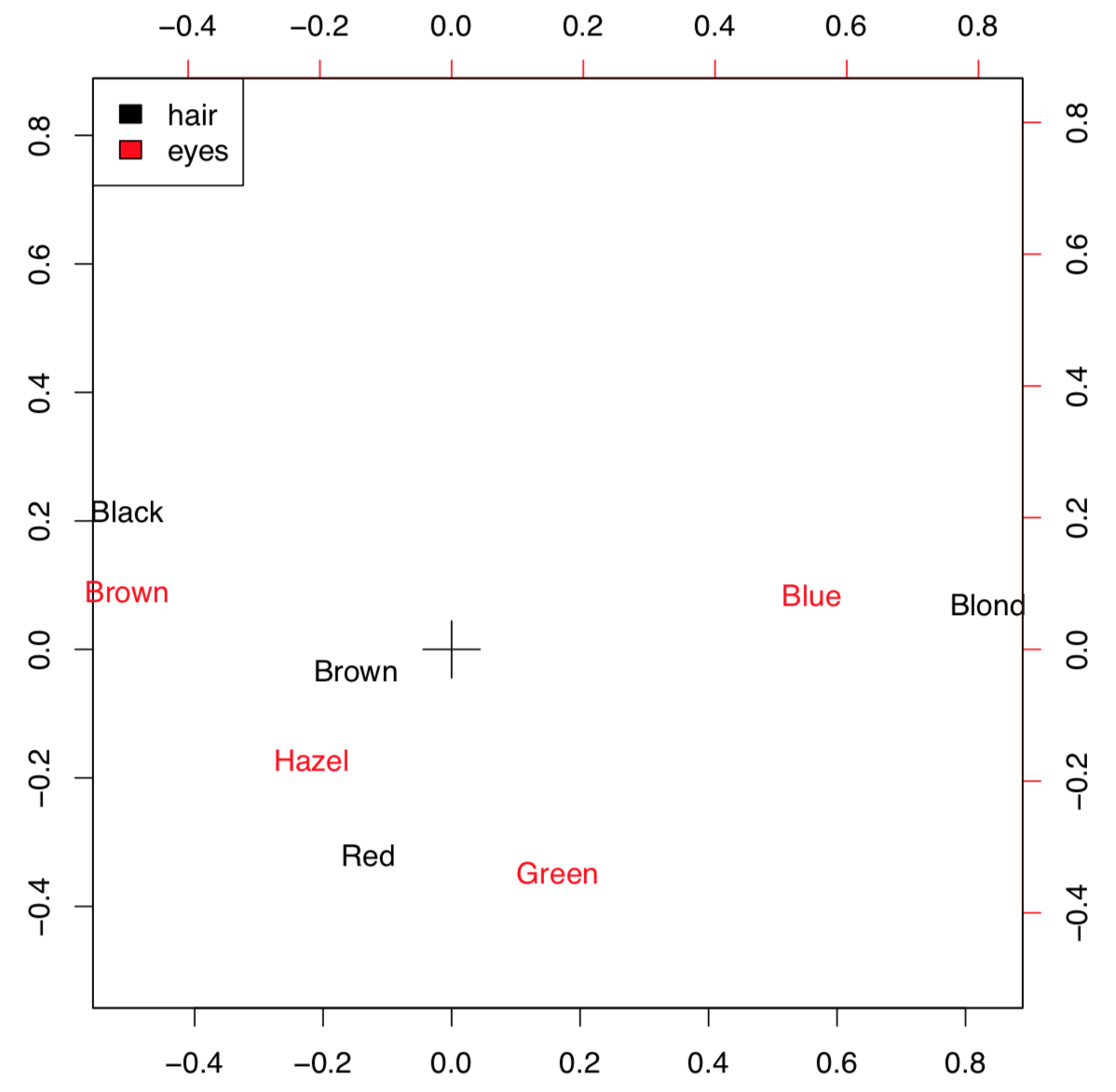
En este ejemplo se utilizan datos de HaireyeColor del capítulo anterior. Trazar visualiza ambos parámetros por lo que si la combinación particular de colores es más frecuente, entonces las posiciones de las palabras correspondientes están más cerca. Por ejemplo, los pelos negros y los ojos marrones suelen aparecer juntos. La posición de estas palabras es más distante del centro (designada con cruz) porque los valores numéricos de estos caracteres son remotos.
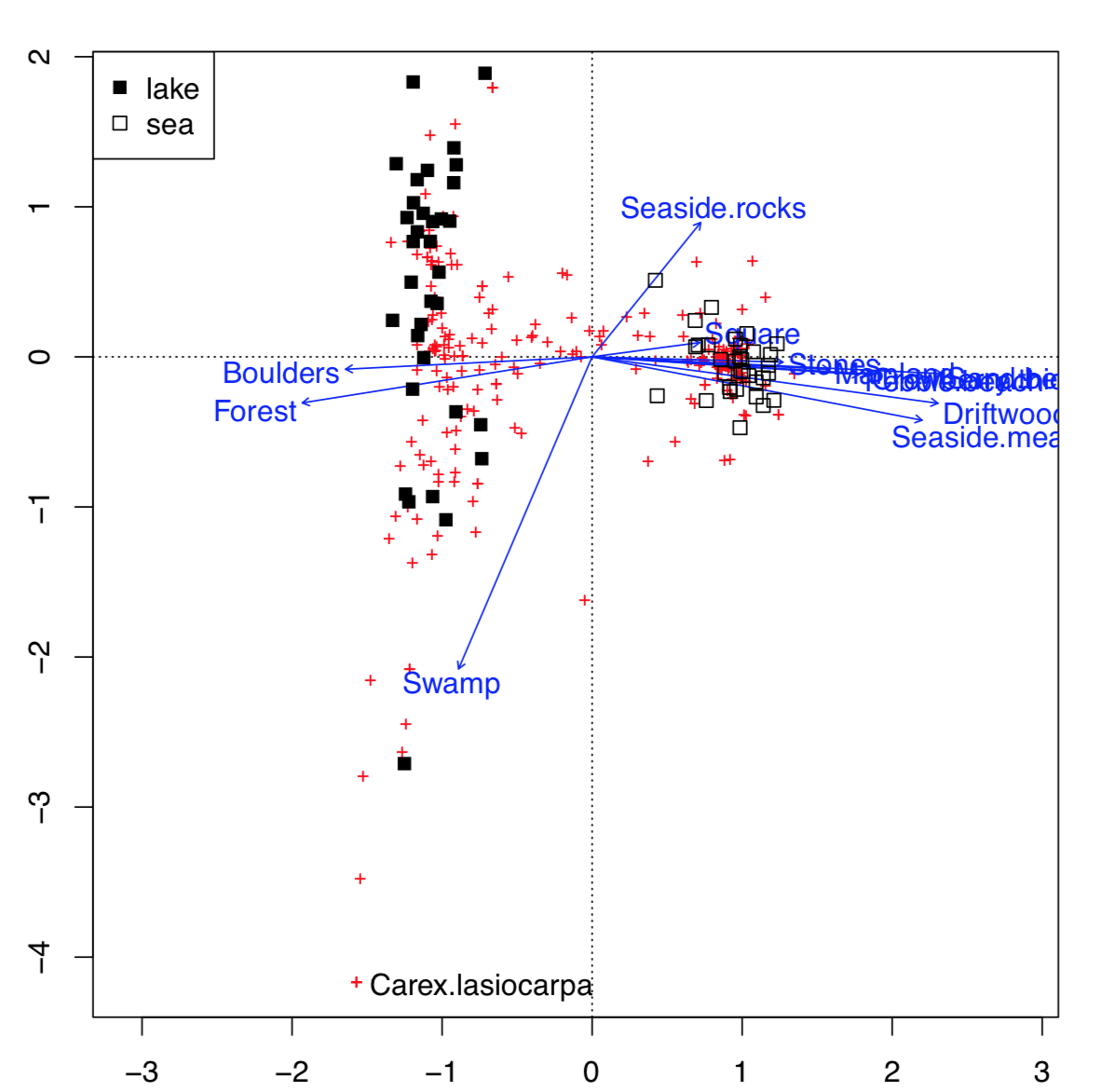
Esta posibilidad de visualizar varios conjuntos de caracteres simultáneamente en una gráfica es la característica impresionante del análisis de correspondencia (Figura\(\PageIndex{11}\)):
Código\(\PageIndex{15}\) (R):
Esto es mucho más avanzado que biplot. Los datos utilizados aquí contenían factores tanto abióticos (ecotopos) como bióticos (especies vegetales), además de la geografía de algunas islas árticas: fueron estas islas lacustres o islas marinas. La parcela fue capaz de organizar todos estos datos: para factores abióticos, utilizó flechas, para biotic—pluses, y para sitios (islas mismas caracterizadas por la suma de todos los factores disponibles, bióticos y abióticos) —cuadrados de diferente color, dependiendo del origen geográfico. Todas las ventajas podrían identificarse con el comando interactivo identify (plot.all.cca, “species”). Lo hicimos solo para una de las especies más destacadas, Carex lasiocarpa (juncia lanosa) que está claramente asociada con islas lacustres, y también con pantanos.
Clasificación con distancias
Una forma importante de clasificación no supervisada es trabajar con distancias en lugar de datos originales. Los métodos basados en la distancia necesitan que primero se calculen las diferencias entre cada par de objetos. La ventaja de estos métodos es que las diferencias podrían calcularse a partir de datos de cualquier tipo: medición, clasificado o nominal.
Distancias
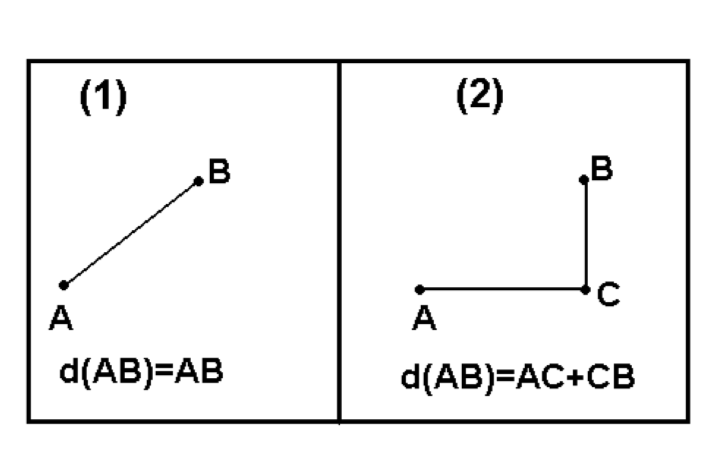
Hay miríadas de formas de calcular la disimilitud (o similitud que es esencialmente la disimilitud inversa)\(^{[2]}\). Una de estas formas ya explicadas anteriormente es una correlación (absoluta inversa). Otras formas populares son la distancia euclidiana (cuadrada) y la distancia de Manhattan (cuadra). Ambos (Figura\(\PageIndex{12}\)) son útiles para variables de medición.
Las distancias de Manhattan son similares a las distancias de conducción, especialmente cuando no hay muchas carreteras disponibles. El siguiente ejemplo son las distancias de conducción entre las ciudades más grandes de Dakota del Norte:
Código\(\PageIndex{16}\) (R):
En la mayoría de los casos, necesitamos convertir variables sin procesar en matriz de distancia. La forma básica es usar dist (). Tenga en cuenta que las variables clasificadas y binarias generalmente requieren diferentes enfoques que se implementan en los paquetes vegan (function vegdist ()) y cluster (function daisy ()). La última función reconoce el tipo de variable y aplica la métrica más adecuada (incluida la distancia universal de Gower); también acepta la métrica especificada por el usuario:
Código\(\PageIndex{17}\) (R):
En biología, se pueden utilizar las distancias taxonómicas de Smirnov, disponibles en el paquete smirnov. En el siguiente ejemplo, utilizamos datos de distribución de especies de plantas en islas pequeñas.
La siguiente trama pretende ayudar al lector a entenderlos mejor. Es solo una especie de mapa que muestra ubicaciones geográficas y tamaños de islas:
Código\(\PageIndex{18}\) (R):
(Por favor, complótelo usted mismo.)
Ahora calcularemos y visualizaremos las distancias de Smirnov:
Código\(\PageIndex{19}\) (R):
Las distancias de Smirnov tienen una característica interesante: en lugar de 0 o 1, la diagonal de la matriz de similitud se rellena con los valores del coeficiente de singularidad (Txx):
Código\(\PageIndex{20}\) (R):
Esto significa que la isla Verik es la más singular en lo que respecta a la ocurrencia de especies de plantas.
Hacer mapas: escalado multidimensional
Hay muchas cosas que hacer con la matriz de distancia. Uno de los más directos es el escalado multidimensional, MDS (el otro nombre es “análisis de coordenadas principales”, PCoA):
Código\(\PageIndex{21}\) (R):
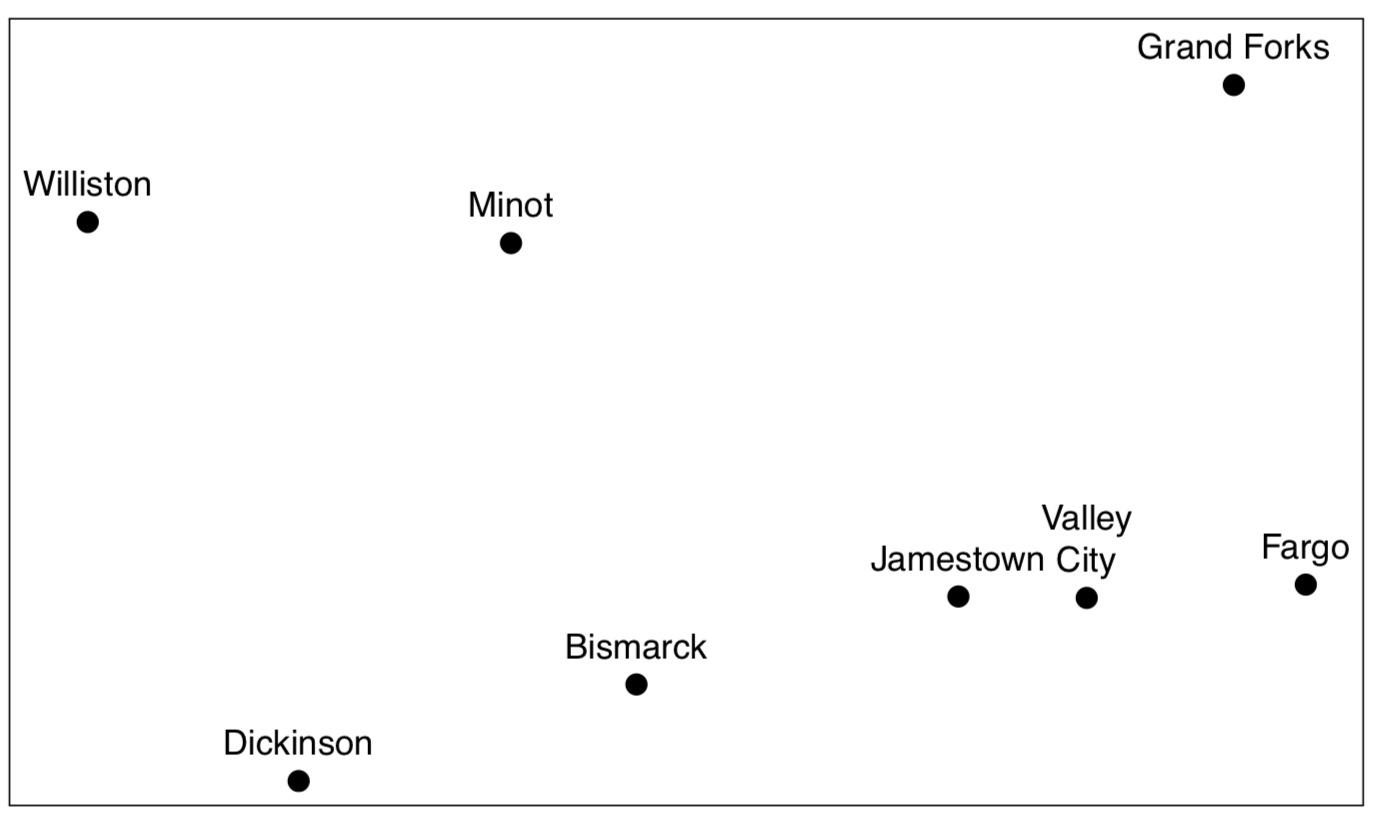
Compara la parcela (Figura\(\PageIndex{13}\)) con cualquier mapa geográfico. Si no tienes un mapa de Dakota del Norte pero tienes estas distancias de manejo, ¡cmdscale () permite recrear el mapa!
Entonces, en esencia, MDS es una tarea inversa a la navegación (encontrar direcciones de manejo desde el mapa): usa “indicaciones de manejo” y hace un mapa a partir de ellas.
Otro ejemplo menos impresionante pero más útil (Figura\(\PageIndex{14}\)) es de los datos brutos de los iris de Fisher:
Código\(\PageIndex{22}\) (R):
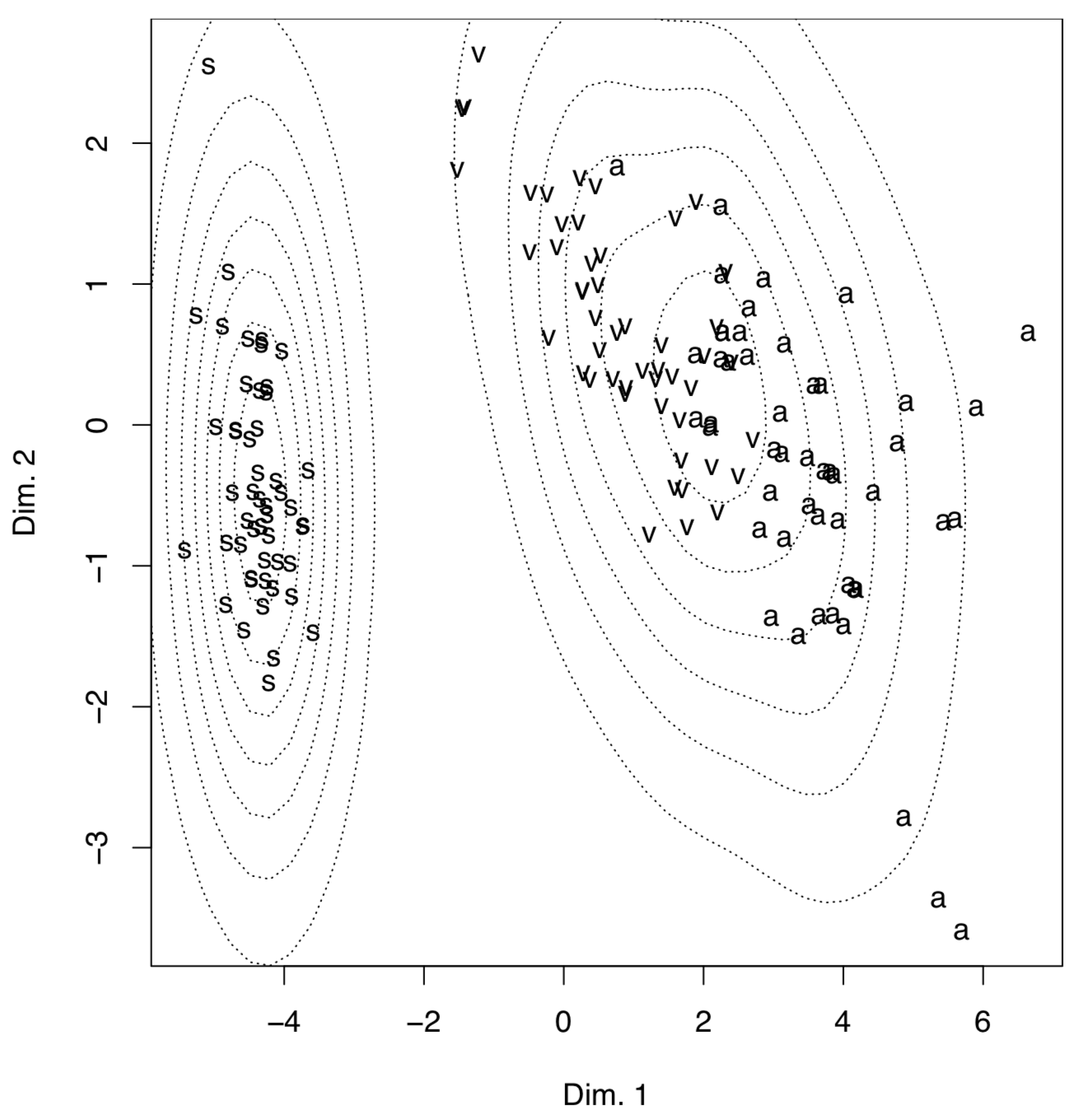
(No hay diferencia real con el PCA porque el escalado multidimensional métrico está relacionado con el análisis de componentes principales; además, la estructura interna de los datos es la misma).
Para hacer la gráfica “más bonita”, agregamos aquí líneas de densidad de cercanía de punto estimadas con la función BKDE2d () a partir del paquete KernSmooth. Otra forma de mostrar la densidad es trazar una superficie 3D como (Figura\(\PageIndex{15}\)):
Código\(\PageIndex{23}\) (R):
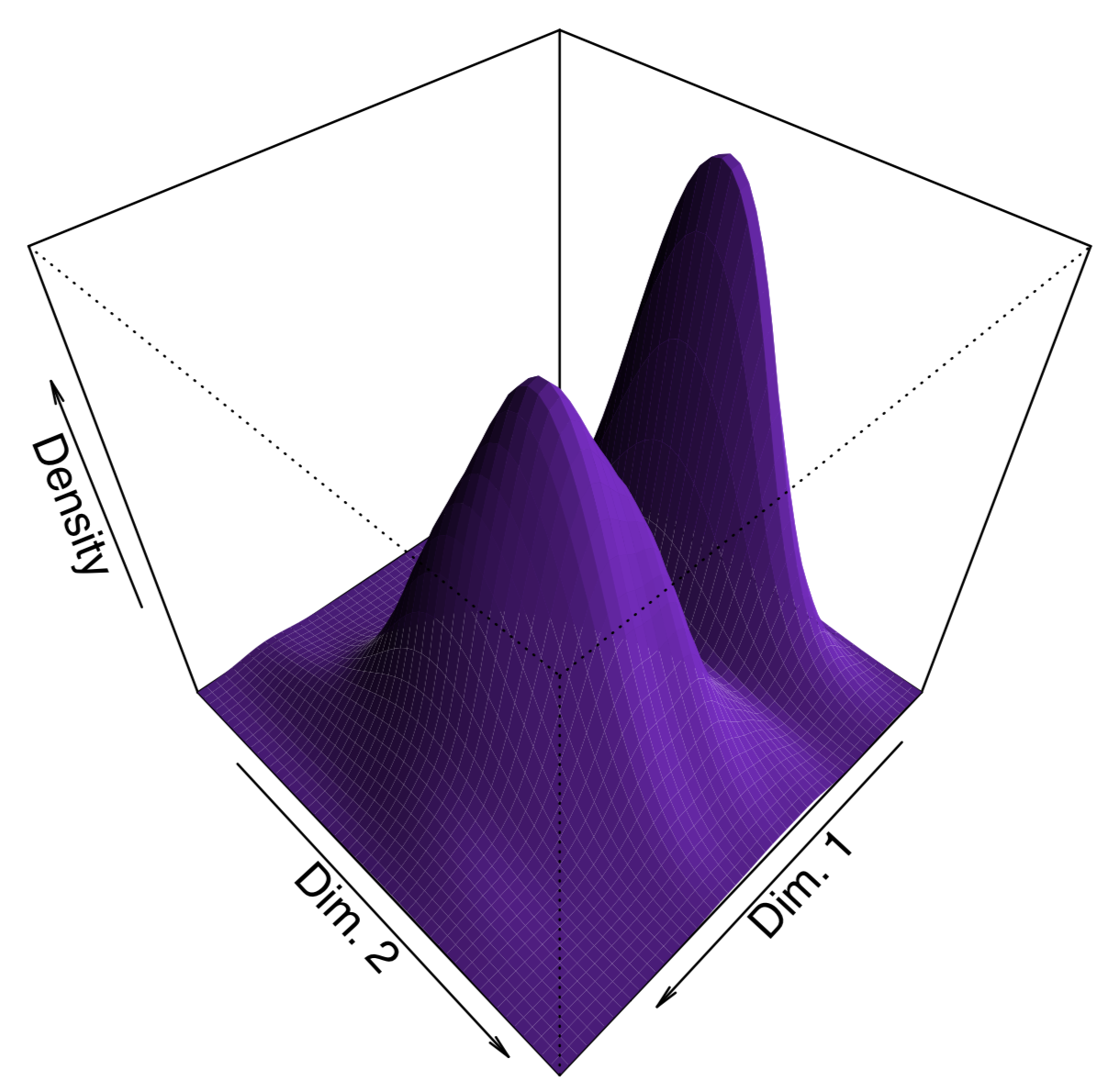
Además de cmdscale (), el paquete MASS (funciones isMDS () y sammon ()) implementa el escalado multidimensional no métrico, y package vegan tiene los metaMDs no métricos avanzados (). El escalado multidimensional no métrico no tiene análogos a las cargas de PCA (importaciones de variables) y proporción de varianza explicada por componente, pero es posible calcular métricas sustitutas:
Código\(\PageIndex{24}\) (R):
En consecuencia (y de manera similar a PCA), el carácter de ancho sépalo influye en la segunda dimensión mucho más que otros tres caracteres. También podemos adivinar que dentro de esta solución no métrica, la primera dimensión toma casi 98% de varianza.
Hacer árboles: agrupación jerárquica
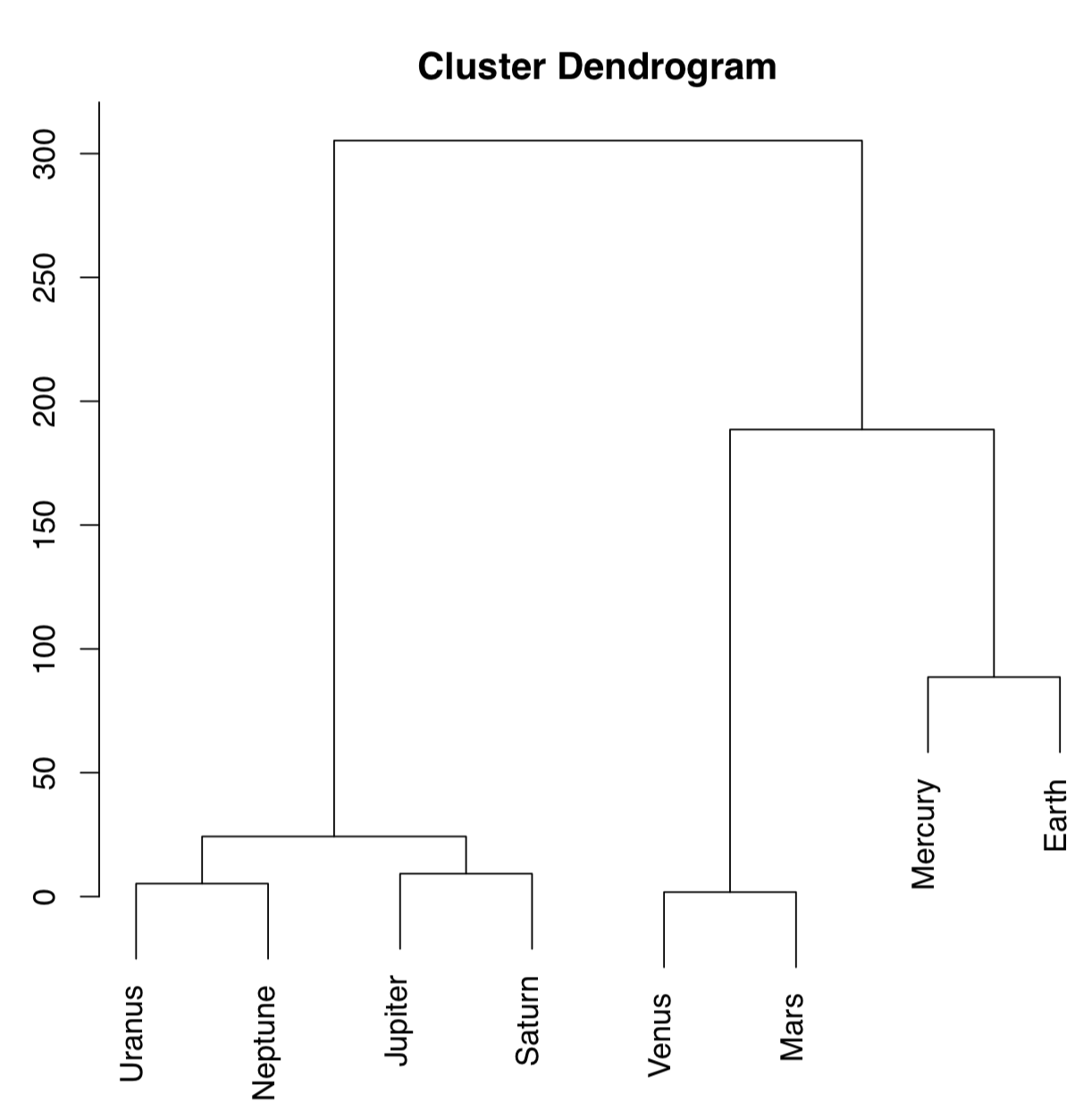
La otra forma de procesar la matriz de distancia es realizar clústeres jerárquicos que producen dendrogramas, o árboles, que son parcelas de “una y media dimensión” (Figura\(\PageIndex{16}\)):
Código\(\PageIndex{25}\) (R):
El método de agrupación de Ward es bien conocido por producir conglomerados nítidos y bien separados (esto, sin embargo, podría llevar a conclusiones falsas si los datos no tienen una estructura aparente). Los planetas distantes son los más similares (en la altura\(\approx25\)), la similitud entre Venus y Marte también es alta (la disimilitud es\(\approx0\)). La Tierra es más sobresaliente, la similitud con Mercurio es menor, en la altura\(\approx100\); pero como Mercurio no tiene verdadera atmósfera, podría ser ignorada.
A partir de esta parcela se podría producir la siguiente clasificación:
- Grupo Tierra: Venus, Marte, Tierra, [Mercurio]
- Grupo Júpiter: Júpiter, Saturno, Urano, Neptuno
En lugar de este enfoque “especulativo”, se puede usar la función cutree () para producir la clasificación explícitamente; esto requiere el objeto hclust () y el número de clústeres deseados:
Código\(\PageIndex{26}\) (R):
Para comprobar qué tan bien realiza la clasificación el método seleccionado, escribimos la función personalizada Misclass (). Esta función calcula la matriz de confusión. Tenga en cuenta que Misclass () asume grupos predichos y observados en el mismo orden, vea también a continuación los resultados de la función fanny ().
La matriz de confusión es una forma sencilla de evaluar el poder predictivo del modelo. Técnica más avanzada del mismo tipo se llama validación cruzada. Como ejemplo, el usuario podría esparcir datos en 10 partes iguales (por ejemplo, con cut ()) y luego, a su vez, hacer que cada parte sea “desconocida” mientras que el resto se convertirá en subconjunto de entrenamiento.
Como puede ver en la tabla, 32% de Iris virginica fueron clasificados erróneamente. El último es posible mejorar, si cambiamos ya sea métrica de distancia, o método de clustering. Por ejemplo, el método de agrupación de Ward da más clústeres separados y tasas de clasificación errónea ligeramente mejores. Por favor pruébalo tú mismo.
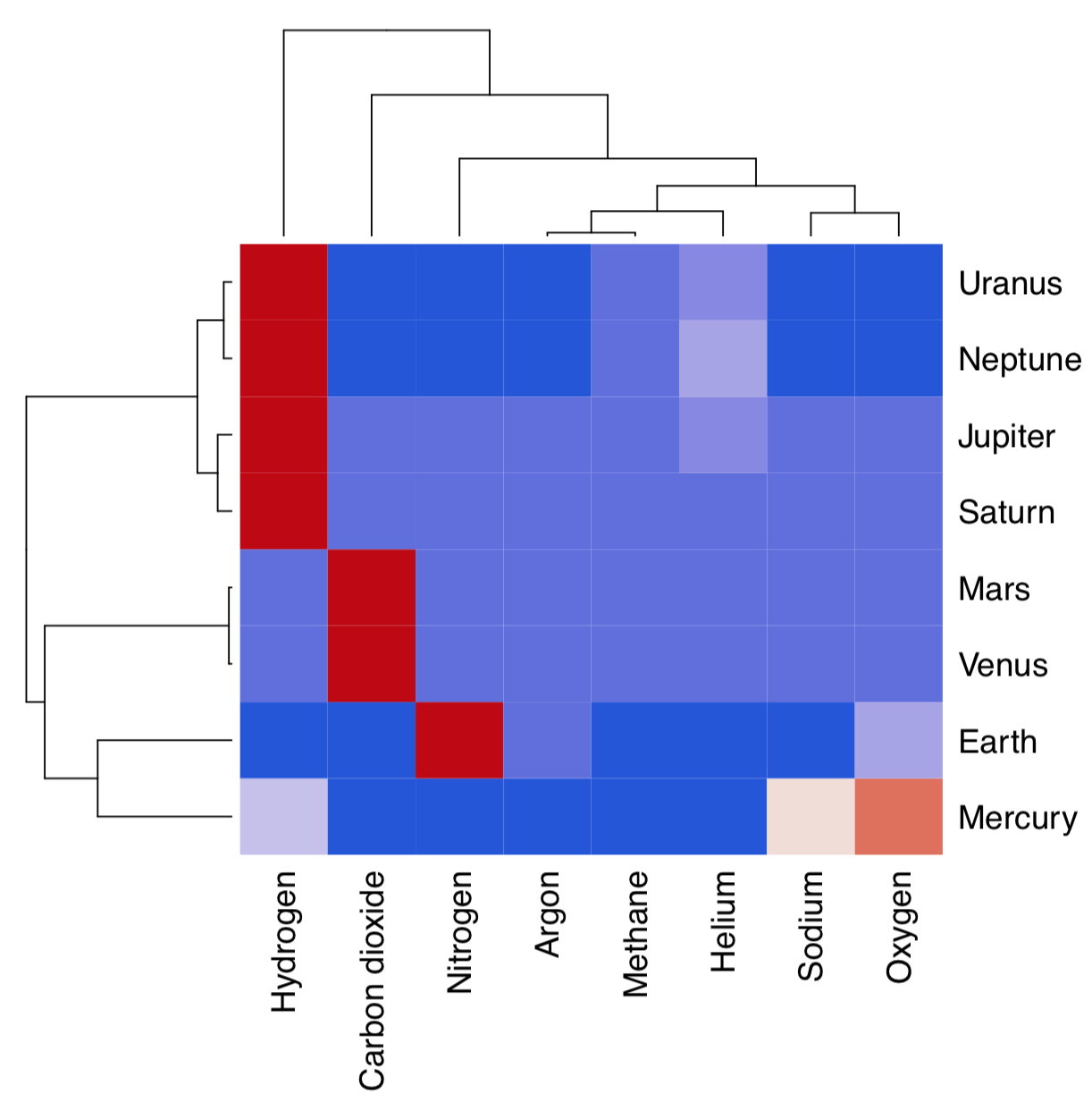
La agrupación jerárquica no devuelve por defecto ninguna importancia de variable. Sin embargo, todavía es posible ayudar a la selección de entidades con el mapa de calor de agrupación (Figura\(\PageIndex{17}\)):
Código\(\PageIndex{27}\) (R):
(Aquí también utilizamos el paquete cetcolor que permite crear paletas de colores perceptualmente uniformes.)
Mapa de calor agrupa por separado filas y columnas y coloca el resultado de la función image () en el centro. Entonces se hace visible qué personajes influyen en qué grupos de objetos y viceversa. En este mapa de calor, por ejemplo, Marte y Venus se agrupan en su mayoría debido a niveles similares de dióxido de carbono.
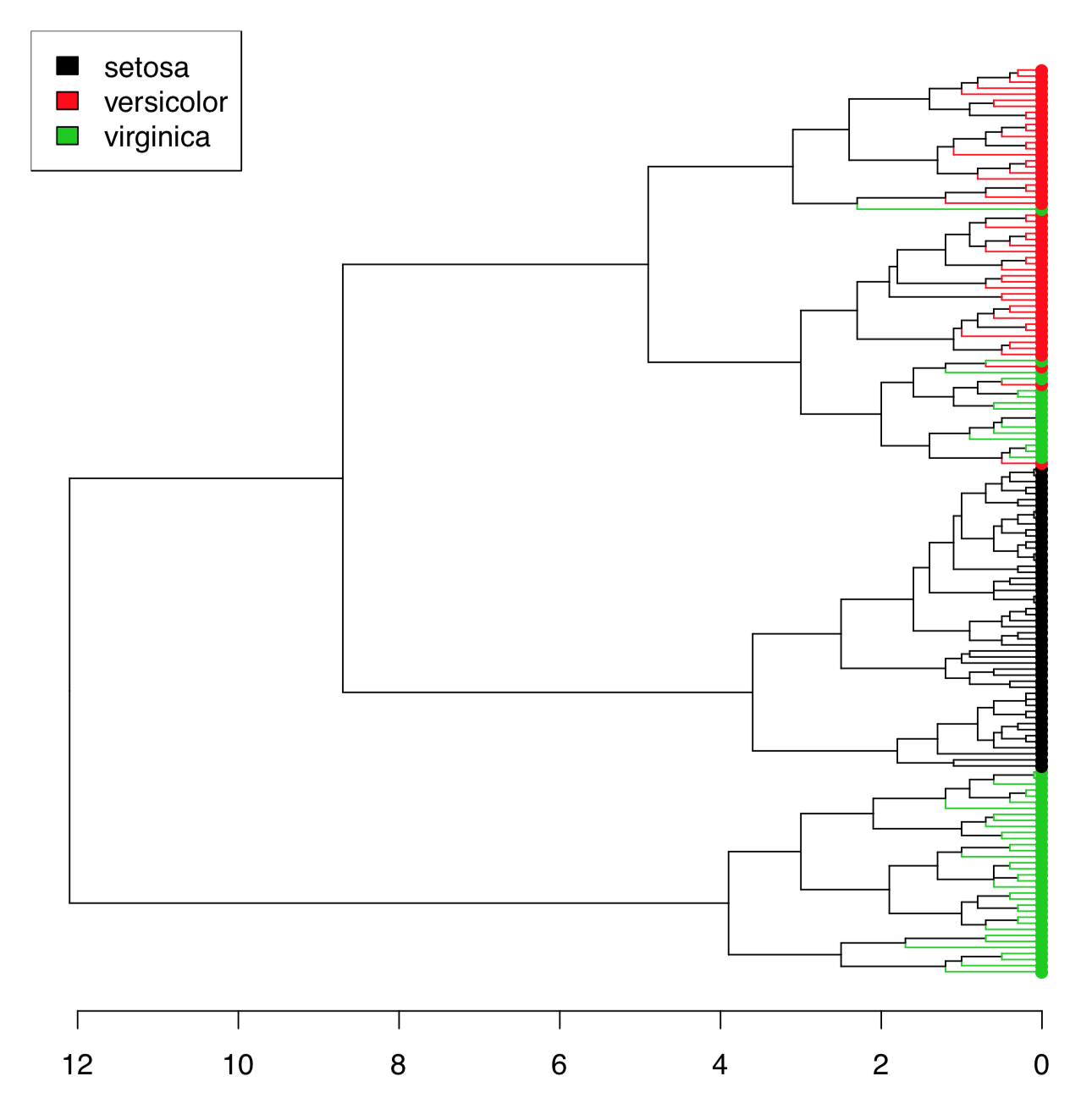
Hay demasiados iris para trazar el dendrograma resultante de la manera común. Una solución alternativa es seleccionar solo algunos iris (ver más abajo). Otro método es usar la función Ploth () (Figura\(\PageIndex{18}\)):
Código\(\PageIndex{28}\) (R):
Ploth () es útil también si uno necesita simplemente rotar el dendrograma. Por favor, compruebe usted mismo lo siguiente:
Código\(\PageIndex{29}\) (R):
(Esto también es una demostración de cómo utilizar la correlación para la distancia. Como verás, la misma conexión entre la ensalada César, el tomate y la enfermedad podría visualizarse con dendrograma. Allí visibles también algunas otras relaciones interesantes.)
archivo.
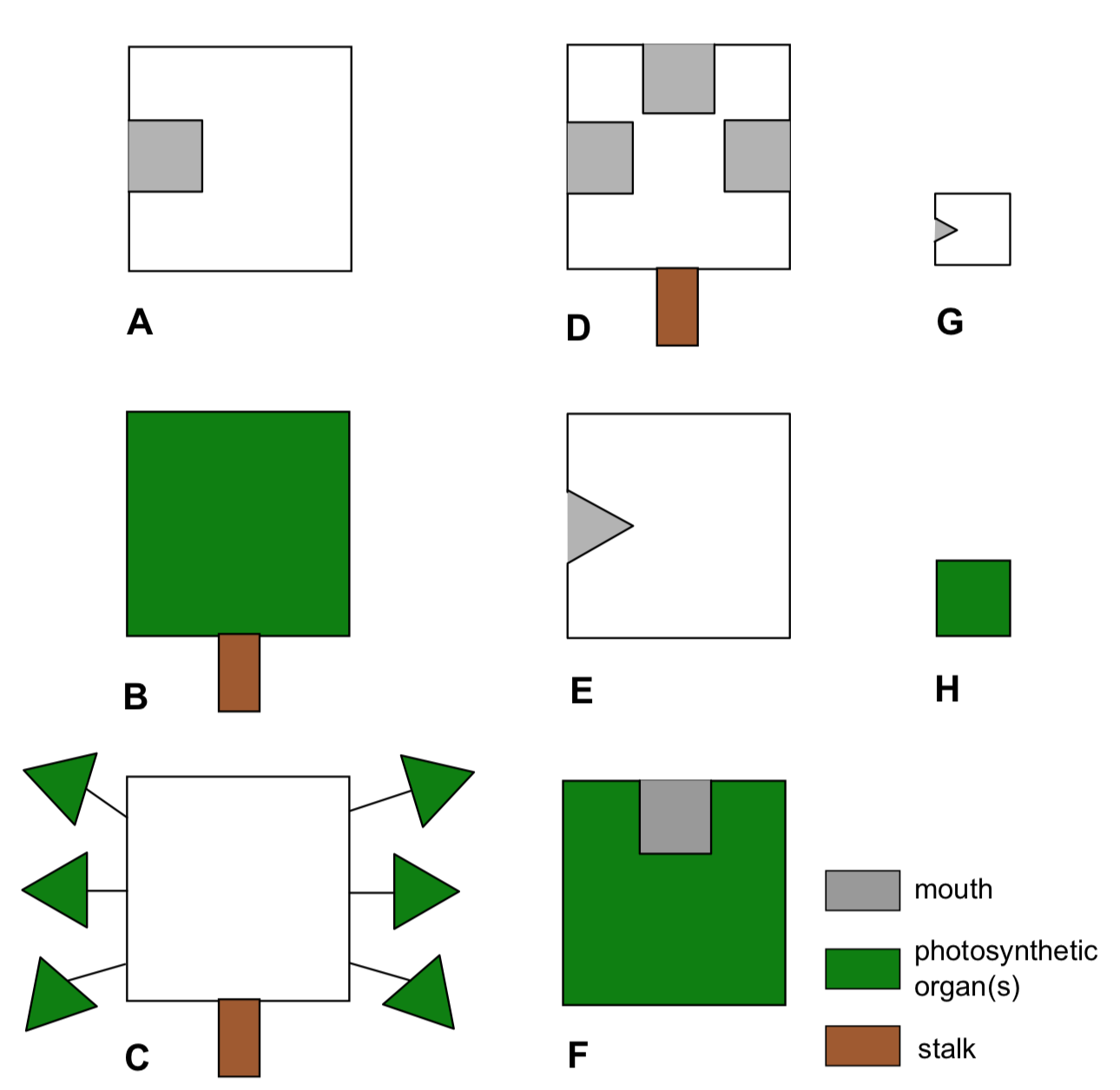
Planet Aqua está completamente cubierto por aguas poco profundas. Este océano está habitado con diversos organismos planos (Figura\(\PageIndex{19}\)). Estas criaturas (las llamamos “kubricks”) pueden fotosintetizar y/o comer otros organismos o sus partes (que coinciden con la forma de sus bocas), y moverse (solo si no tienen tallos). Proporcionar el dendrograma para especies de kubrick basado en el resultado del agrupamiento jerárquico.
Cómo conocer el mejor método de clustering
El análisis jerárquico de conglomerados y los parientes (por ejemplo, árboles de filogenia) son visualmente atractivos, pero hay tres preguntas importantes que deben resolverse: (1) qué distancia es la mejor (esto también es relevante para otros métodos basados en la distancia); (2) qué método de agrupación jerárquica es el mejor; y (3) cómo evaluar la estabilidad de los racimos.
La segunda pregunta es relativamente fácil de responder. La función Co.test (dist, tree) de asmisc.r revela consistencia entre objeto de distancia y clusterización jerárquica. Es esencialmente una prueba de correlación entre las distancias iniciales y las distancias reveladas a partir de la estructura cofenética del dendrograma.
Las distancias cofenéticas son útiles de muchas maneras. Por ejemplo, para elegir el mejor método de clusterización y, por lo tanto, responder a la segunda pregunta, se podría usar
Código\(\PageIndex{30}\) (R):
(Haz y revisa esta parcela tú mismo. ¿Qué clustering es mejor?)
Tenga en cuenta, sin embargo, estas “mejores” puntuaciones no siempre son las mejores para usted. Por ejemplo, todavía se podría decidir usar Ward.d porque hace que los racimos sean nítidos y separados visualmente.
Para elegir el mejor método de distancia, uno podría usar el enfoque visualmente similar:
Código\(\PageIndex{31}\) (R):
(Nuevamente, por favor revise la parcela usted mismo.)
De hecho, solo visualiza la correlación entre el escalado multidimensional de distancias y el análisis de componentes principales de datos brutos. Sin embargo, sigue siendo útil.
Cómo comparar clústeres
La agrupación jerárquica son dendrogramas y no es fácil compararlos “fuera de la caja”. Varios métodos diferentes permiten comparar dos árboles.
Podemos emplear métodos asociados a filogenias biológicas (estos árboles son esencialmente dendrogramas).
Supongamos que hay dos agrupamientos:
Código\(\PageIndex{32}\) (R):
Library ape tiene la función dist.topo () que calcula la distancia topológica entre árboles, y la biblioteca phangorn calcula varios de esos índices:
Código\(\PageIndex{33}\) (R):
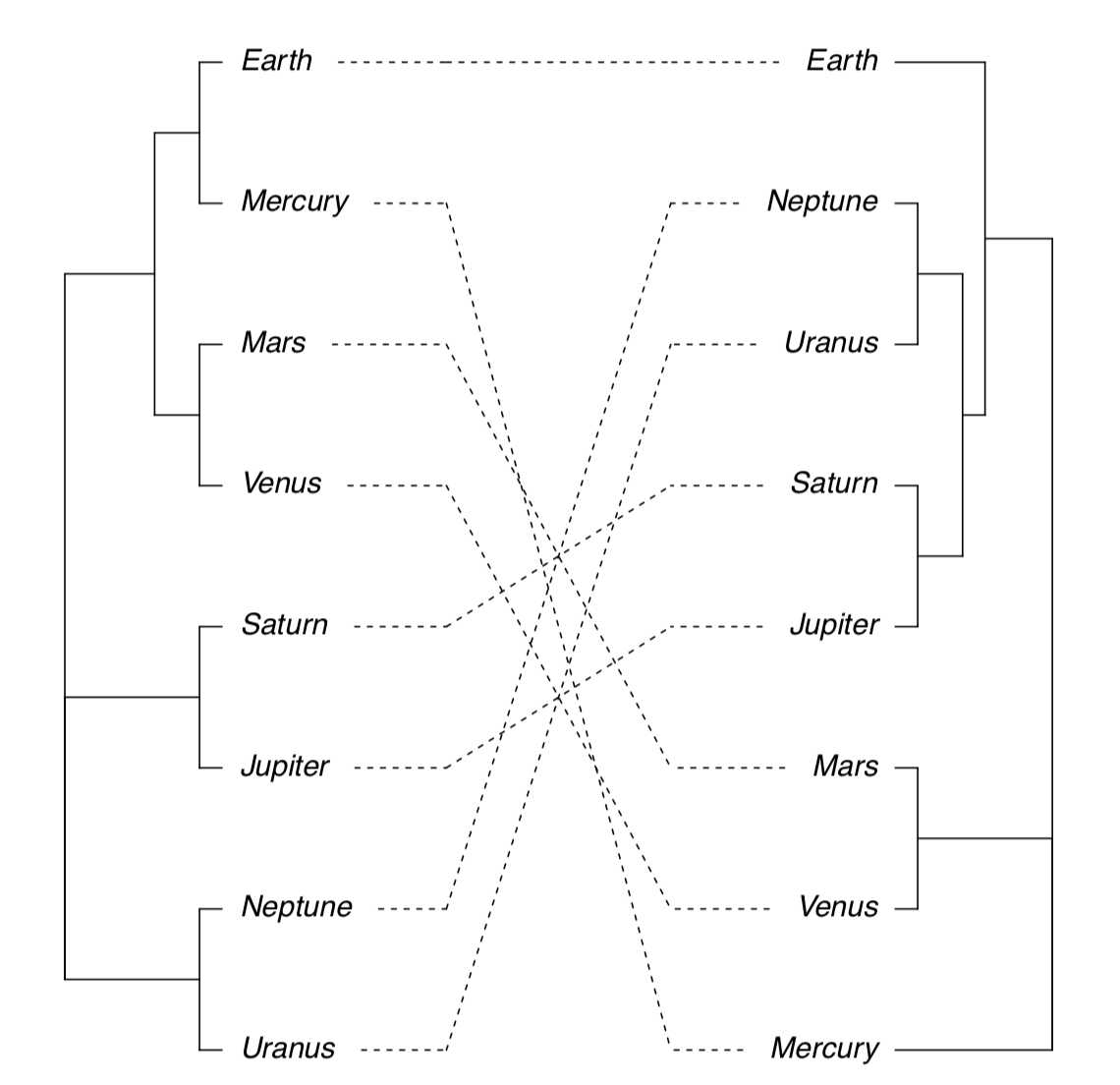
La siguiente posibilidad es trazar dos árboles uno al lado del otro y mostrar diferencias con líneas que conectan las mismas puntas (Figura\(\PageIndex{20}\)):
Código\(\PageIndex{34}\) (R):
(Tenga en cuenta que a veces es posible que necesite rotar la rama con la función rotate (). La rotación no cambia el dendrograma).
También es posible trazar un árbol de consenso que muestra solo aquellos clústeres que aparecen en ambos agrupamientos:
Código\(\PageIndex{35}\) (R):
(Por favor, haga esta parcela usted mismo.)
El mapa de calor también podría usarse para visualizar similitudes entre dos dendrogramas:
Código\(\PageIndex{36}\) (R):
(hclust.match () cuenta coincidencias entre dos dendrogramas (que se basan en los mismos datos) y luego heatmap () traza estos recuentos como colores, y también suministra la configuración de consenso como dos dendrogramas idénticos en la parte superior y en la izquierda. Por favor, haz esta parcela tú mismo.)
Tanto el escalado multidimensional como el agrupamiento jerárquico son métodos basados en la distancia. Por favor, haga y revise la siguiente parcela (del paquete vegan3d) para entender cómo compararlos:
Código\(\PageIndex{37}\) (R):
Qué tan buenos son los clústeres resultantes
Hay varias formas de verificar qué tan buenos son los clústeres resultantes, y muchas se basan en la replicación de arranque (ver Apéndice).
La función Jclust () presenta un método para arrancar biparticiones y trazar árbol de consenso con valores de soporte (Figura\(\PageIndex{21}\):
Código\(\PageIndex{38}\) (R):
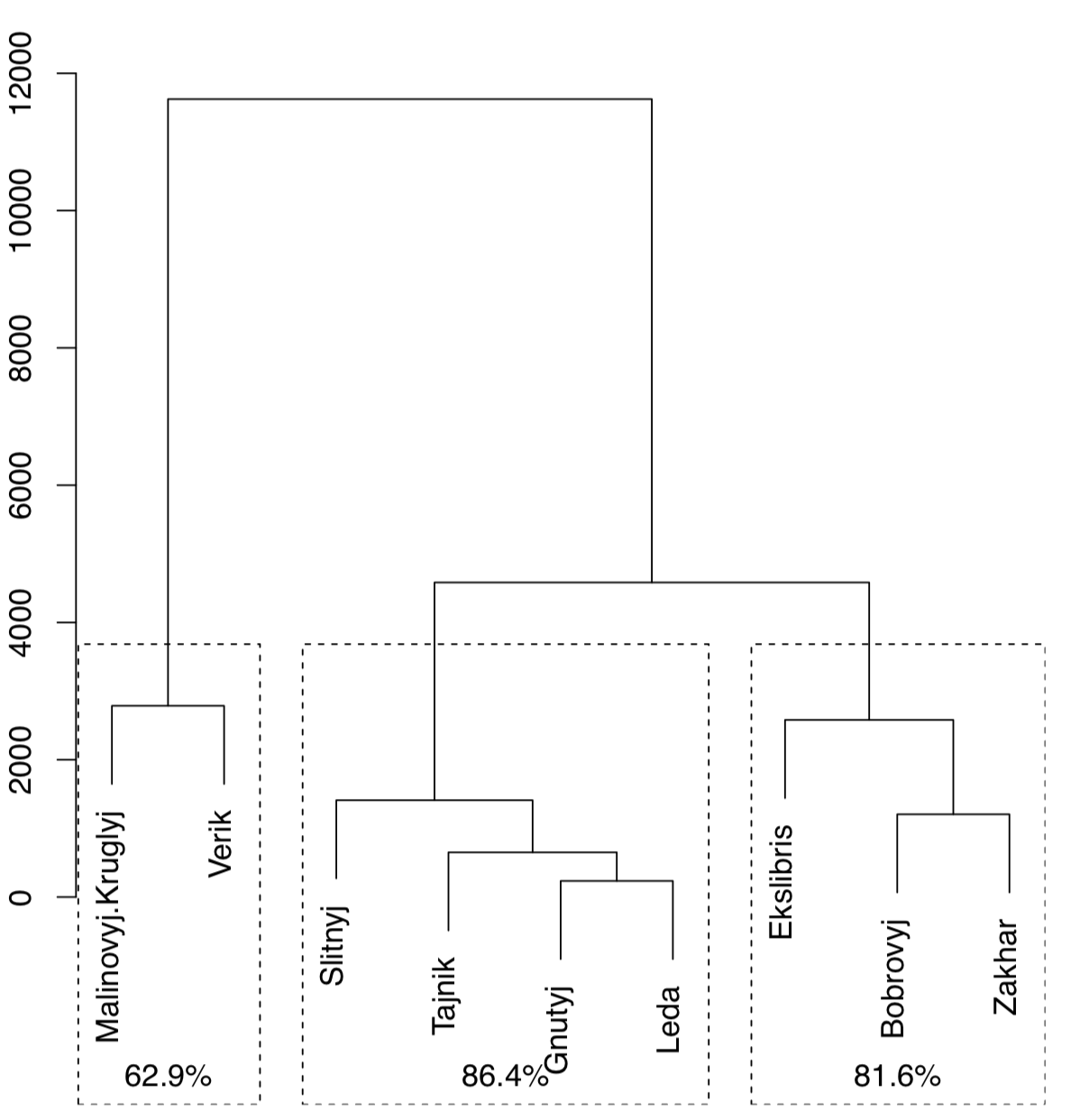
(Tenga en cuenta que Jclust () usa cutree () y por lo tanto funciona solo si “conoce” el número de clústeres deseados. Dado que el resultado del consenso se relaciona con el número de conglomerados, las parcelas con diferentes números de conglomerados serán diferentes).
Otra forma es usar el paquete pvclust que tiene la capacidad de calcular el soporte para clústeres a través de bootstrap (Figura\(\PageIndex{22}\)):
Código\(\PageIndex{39}\) (R):
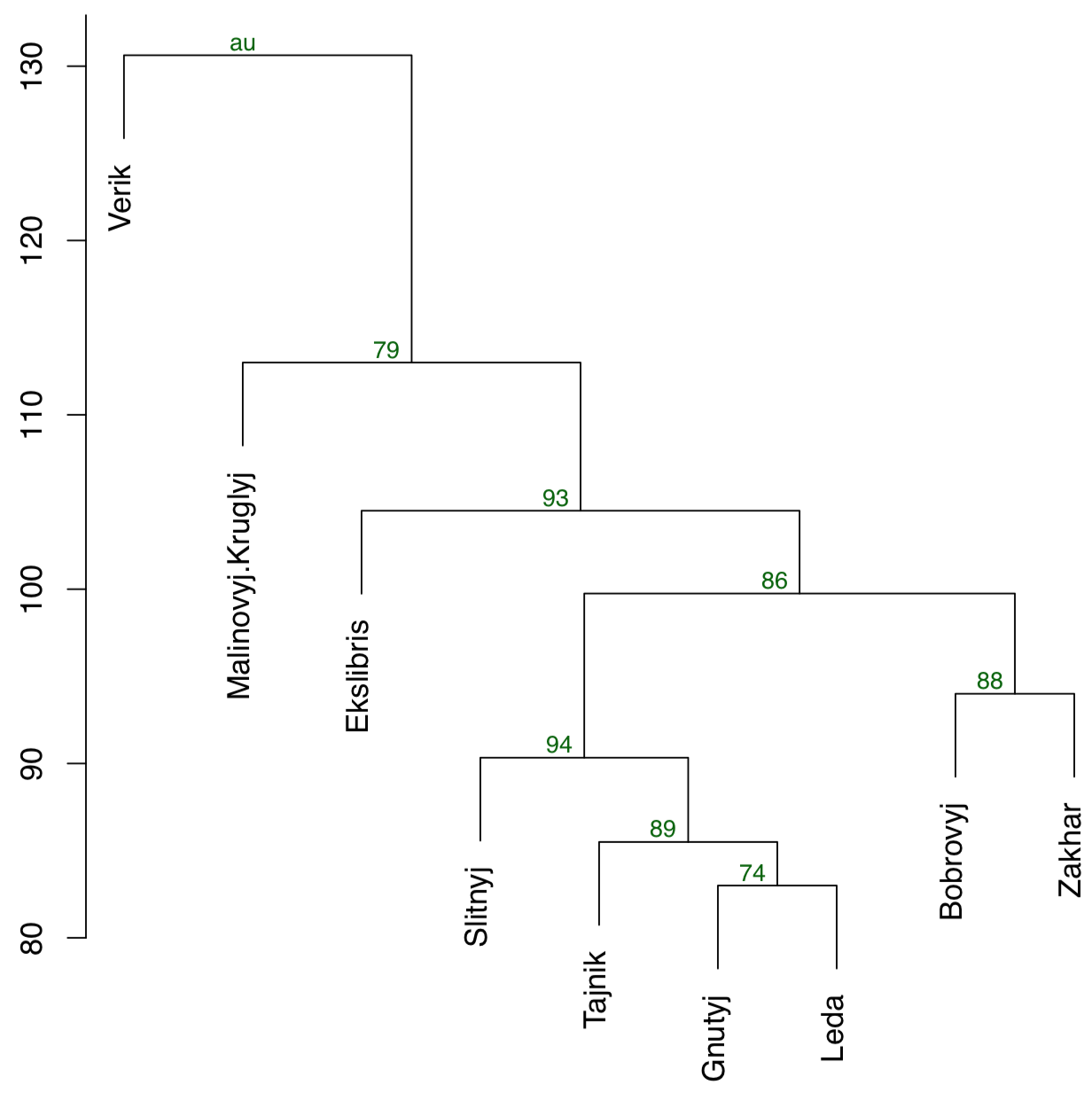
(La función pvclust () agrupa columnas, no filas, así que tenemos que transponer datos de nuevo. En la parcela, los valores numéricos de estabilidad de conglomerados (au) se ubican por encima de cada nodo. Cuanto más cerca estén estos valores a 100, mejor.)
También hay la función booTA () en el conjunto asmisc.r que permite bootstrap clustering con métodos del paquete filogenético ape:
Código\(\PageIndex{40}\) (R):
(Este método requiere hacer una función anónima que utilice los métodos que desee. También traza tanto el árbol de consenso (sin valores de soporte) como el árbol original con valores de soporte. Por favor, haz estos árboles. Tenga en cuenta que por defecto, solo se muestran valores de soporte mayores a 50%.)
Hacer grupos: k-means y amigos
Aparte de jerárquico, hay muchas otras formas de agrupación. Por lo general, no devuelven ninguna ordenación (“mapa”) y proporcionan solo membresía al clúster. Por ejemplo, k-means clustering intenta obtener el número de clústeres especificado a priori a partir de los datos brutos (no necesita que se suministre la matriz de distancia):
Código\(\PageIndex{41}\) (R):
La agrupación de K-medias no traza árboles; en su lugar, para cada objeto devuelve el número de su clúster:
Código\(\PageIndex{42}\) (R):
(Como ve, los errores de clasificación errónea son bajos).
En lugar de un número de clúster a priori, la función kmeans () también acepta números de fila de centros de clúster.
La agrupación espectral del paquete kernlab es un método superficialmente similar capaz de separar elementos realmente enredados:
Código\(\PageIndex{43}\) (R):

Los métodos del kernel (como la agrupación espectral) recalculan los datos primarios para que sean más adecuados para el análisis. Soporte de máquinas vectoriales (SVM, ver abajo) es otro ejemplo. También hay kernel PCA (función kpca () en el paquete kernlab).
El siguiente grupo de métodos de agrupamiento se basa en la lógica difusa y toma en cuenta la borrosidad de las relaciones. Siempre existe la posibilidad de que un objeto particular clasificado en el clúster A pertenezca al clúster B diferente, y la agrupación difusa intenta medir esta posibilidad:
Código\(\PageIndex{44}\) (R):
La parte textual de la salida de fanny () es lo más interesante. Cada fila contiene múltiples valores de pertenencia que representan la probabilidad de que este objeto esté en el clúster en particular. Por ejemplo, la sexta planta probablemente pertenece al primer racimo pero también hay atracción visible hacia el tercer racimo. Además, fanny () puede redondear membresías y producir clústeres duros como otros métodos de clúster:
Código\(\PageIndex{45}\) (R):
(Tuvimos que volver a nivelar la variable Especie porque fanny () da el número 2 al racimo de Iris virginica).
Cómo conocer los números de clúster
Todos los métodos “k-means and friends” quieren saber el número de clústeres antes de que comiencen. Entonces, ¿cómo saber a priori cuántos clusters presentan en los datos? Esta pregunta es una de las más importantes en la agrupación, tanto práctica como teóricamente.
El análisis visual de la trama de pancartas (inventado por Kaufman & Rousseeuw, 1990) podría predecir este número (Figura\(\PageIndex{24}\)):
Código\(\PageIndex{46}\) (R):
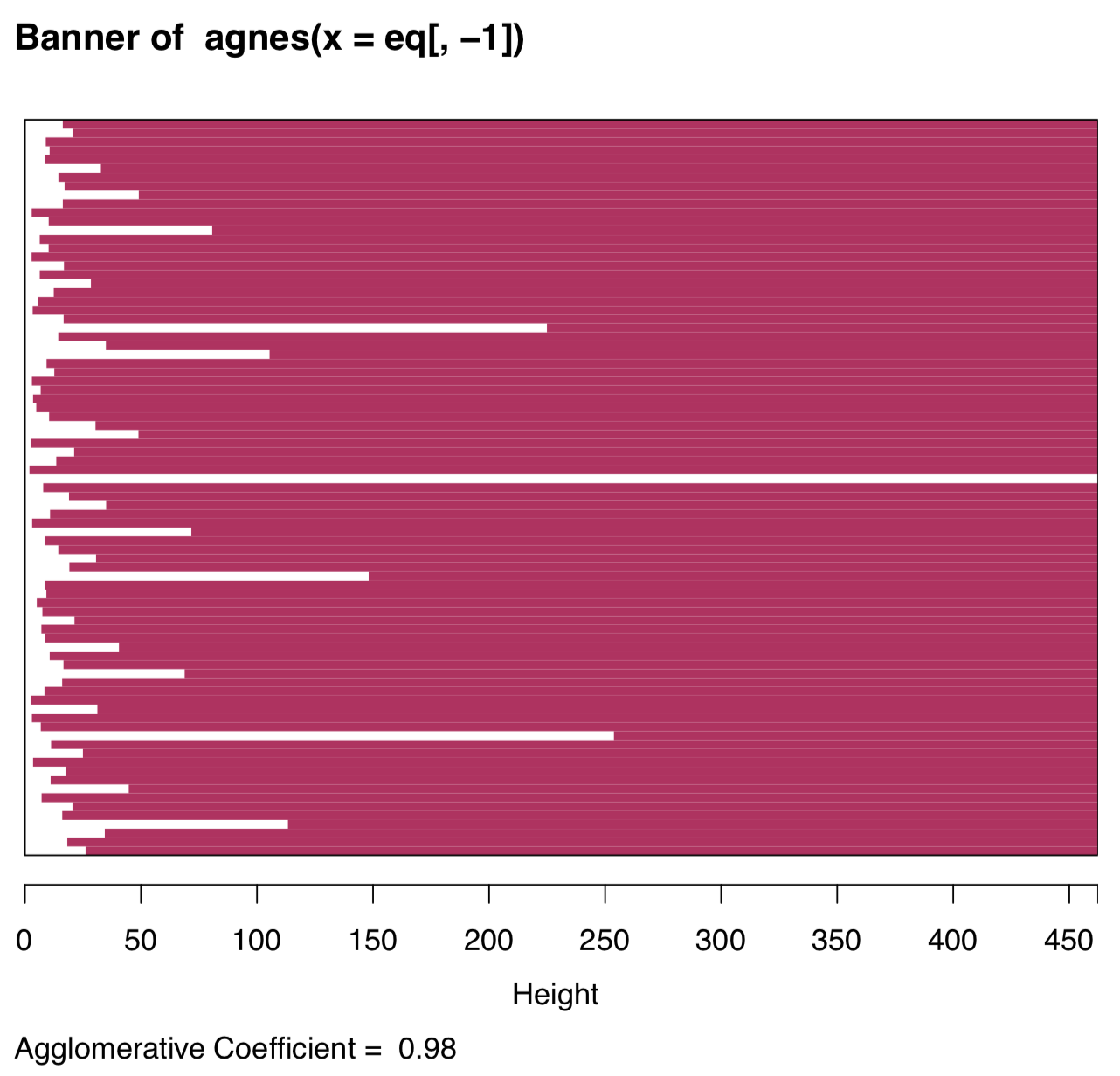
Las barras blancas a la izquierda representan datos no agrupados, las líneas granate a la derecha muestran la altura de posibles cúmulos. Por lo tanto, dos clusters es la solución más natural, cuatro clusters deberían ser la siguiente opción posible.
La agrupación basada en modelos permite determinar cuántos clústeres están presentes en los datos y también la pertenencia a clústeres. El método asume que los clústeres tienen la naturaleza particular y las formas multidimensionales:
Código\(\PageIndex{47}\) (R):
(Como veis, revela sólo dos cúmulos. Esto es explicable porque en los datos del iris dos especies son mucho más similares que la tercera.)
DBSCAN es el potente algoritmo para el big data (como imágenes ráster que constan de miles de millones de píxeles) y existe el paquete R con el mismo nombre (en minúsculas). DBSCAN revela cuántos clústeres hay en los datos a una resolución particular:
Código\(\PageIndex{48}\) (R):
(No se muestran las parcelas, por favor hágalo usted mismo. Primera parcela ayuda a encontrar el tamaño de barrio (mirada en la rodilla). El segundo ilustra los resultados. De manera similar a la agrupación basada en modelos, DBSCAN revela por defecto solo dos clústeres en los datos del iris).
Tenga en cuenta que si bien DBSCAN no pudo recuperar las tres especies, recuperó nubes, y también coloca puntos marginales en el grupo de “ruido”. DBSCAN, como ves, es útil para suavizar, parte importante del reconocimiento de imágenes. El parámetro eps permite cambiar la “resolución” de la agrupación y encontrar más, o menos, clústeres. DBSCAN se relaciona con T-SNE (ver arriba) y con métodos supervisados basados en proximidad (como KnN, ver abajo). También puede supervisarse a sí mismo y predecir clústeres para nuevos puntos. Tenga en cuenta que k-medias y DBSCAN se basan en proximidades calculadas específicamente, no directamente en distancias.
Las estrellas de datos contienen información sobre las 50 estrellas más brillantes en el cielo nocturno, su ubicación y constelaciones. Por favor, use DBSCAN para hacer constelaciones artificiales en la base de la proximidad de estrellas. ¿Cómo se relacionan con las constelaciones reales?
Obsérvese que la ubicación (ascensión derecha y declinación) se da en grados u horas (sistema sexagesimal), deben convertirse en decimales.
El método de “cambio de media” busca modos dentro de los datos, que en esencia, es similar a encontrar proximidades. El algoritmo de desplazamiento medio del núcleo es lento, por lo que la versión aproximada de “desenfoque” es típicamente preferible:
Código\(\PageIndex{49}\) (R):
Otro enfoque para encontrar el número de clúster es similar a la trama de pantalla de PCA:
Código\(\PageIndex{50}\) (R):
(Por favor revise esta parcela usted mismo. Al igual que en la parcela de estandarte, es visible que los “acantilados” relativos más altos están después de los números de racimo 1 y 4
Colección asmisc.r contiene la función Peaks () que ayuda a encontrar máximos locales en una secuencia de datos simple. El número de estos picos en el histograma (con el número sensible de descansos) debe apuntar al número de clústeres:
Código\(\PageIndex{51}\) (R):
>
[1] 3(“Tres” es el primer número de picos después de “uno” y no cambia cuando 8 < se rompe < 22.)
Finalmente, el paquete integrador NBCLust permite utilizar diversos métodos para evaluar el número putativo de clústeres:
Código\(\PageIndex{52}\) (R):
Cómo comparar diferentes ordenaciones
La mayoría de los métodos de clasificación dan como resultado alguna ordenación, gráfica 2D que incluye todos los puntos de datos. Esto permite compararlos con el análisis de Procrustes (ver Apéndice para más detalles) que rota y escala una matriz de datos para hacer en máxima similar con la segunda (objetivo). Comparemos los resultados de los clásicos PCA y T-SNE:
Código\(\PageIndex{53}\) (R):
La gráfica resultante (Figura 7.3.1) muestra qué tan densos son los puntos en t-SNE y cómo los propaga PCA. ¿Cuál de los métodos hace mejor agrupación? Encuéntralo tú mismo.
Referencias
1. Paquete Boruta es especialmente dios para toda la selección de características relevantes.