
9.3: Preniltransferasa de proteínas - una sustitución híbrida SN1-SN2
- Page ID
- 2379

9.3A: The biological relevance of the protein prenyltransferase reaction
Although it is instructive to consider examples of nucleophilic substitution reactions that are thought to occur by completely SN1 or completely SN2 mechanisms, the reality is that many biologically relevant substitution reactions fall somewhere in between these two extremes. A good example of this is the reaction catalyzed by an enzyme called protein prenyltransferase:
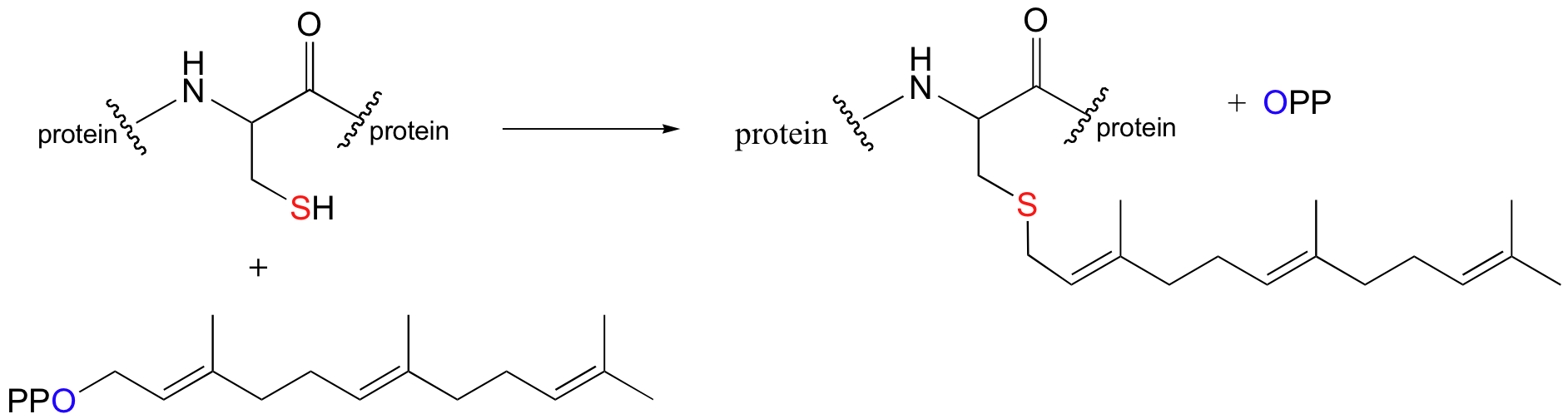
In this nucleophilic substitution reaction, a cysteine residue on certain proteins is modified with a branched-chain 'isoprenyl' hydrocarbon. Do not be confused by the fact there are two proteins involved in this reaction - one is the catalyst (the enzyme, protein prenyltransferase), and the second is a substrate in the reaction.
This reaction, and the chemical mechanism involved, is of particular interest to biomedical scientists. Many of the proteins that are substrates for this type of modification are involved in cell signaling processes, and they are not able to carry out their biological functions unless they are anchored to a cell’s lipid bilayer membrane. This is the role played by the isoprenyl group (see section 2.4B for a description of the molecular structure of lipid bilayer membranes).
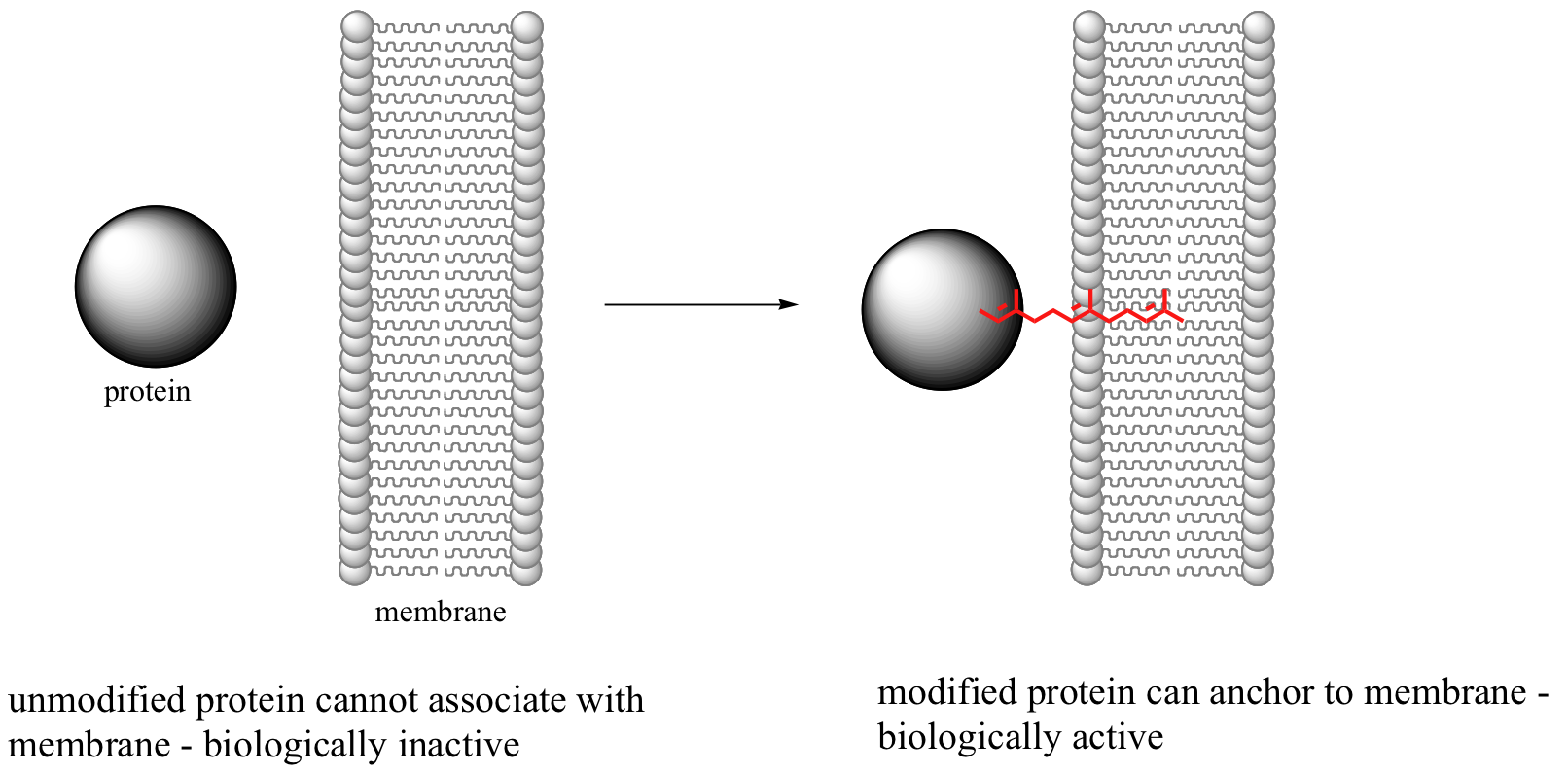
Some proteins that require this isoprenyl anchor attachment have been implicated in tumor formation. Scientists hope that if they can find a way to shut down the prenylation reaction, the tumor-causing proteins will not be able to anchor to cell membranes and thus will remain inactive. The search is on for an effective inhibitor of this enzyme to serve as a potential anti-tumor drug.
Experimental evidence (see section 9.3C) indicates that when the substrate protein is bound to the active site of the enzyme, the cysteine is in a deprotonated state (a thiolate ion). A thiolate is a very potent nucleophile! Notice that the leaving group in this reaction is inorganic pyrophosphate.
9.3B: Determining the mechanism of protein prenyltransferase with fluorinated substrate analogs
Scientists are very interested in understanding the details of the protein prenyltransferase reaction. One question that has been successfully addressed is whether the mechanism is SN1, SN2, or somewhere in between. Researchers measured the rate of the reaction with the normal isoprenyl substrate, and also with two synthetic derivatives in which nearby methyl hydrogens were replaced with either one or three fluorines (Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 5008).
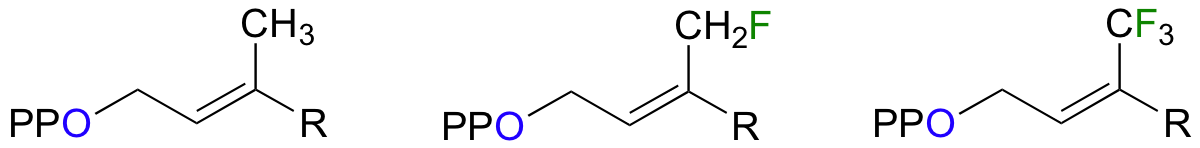
Fluorine has an atomic radius that is close to that of hydrogen, and thus a hydrogen-to-fluorine substitution does not effect the sterics of the reaction to a great extent. However, as you well know, fluorine is very different from hydrogen in terms of electronics: fluorine is highly electronegative (in other words, highly electron-withdrawing). If the reaction is SN1, it should proceed through a positively-charged intermediate: this intermediate would be an allylic carbocation (see section 8.4B) , with the positive charge delocalized by resonance.
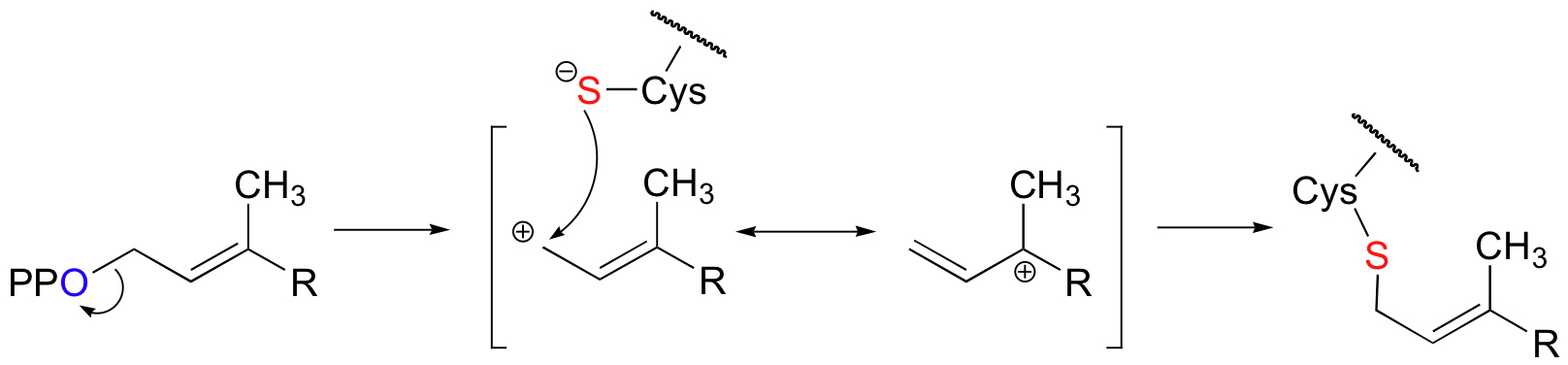
Fluorines, being powerful electron-withdrawing groups, would significantly destabilize this hypothetical carbocation intermediate, and thus would slow down the first (rate-determining) step of an SN1 reaction.
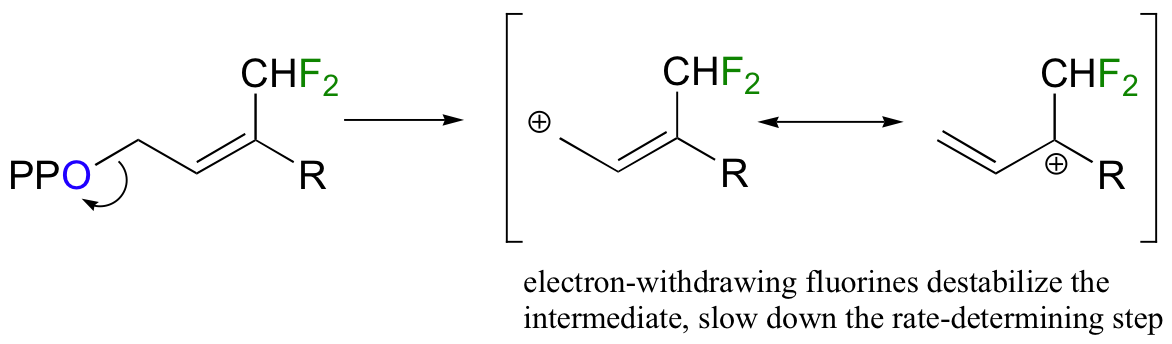
Thus, it stands to reason that the overall rate of an SN1 reaction would decrease each time a fluorine substitution is made, and that the slowest reaction rate would be with the trifluorinated substrate.
Conversely, if the reaction is SN2, the fluorines should not slow down the rate of the reaction because there is no carbocation intermediate to destabilize.
This experimental premise is supported by similar experiments with model non-enzymatic substitution reactions that are known to proceed by SN1 or SN2 mechanisms. As expected for the SN1 model reaction, adding fluorines slowed down the reaction considerably.
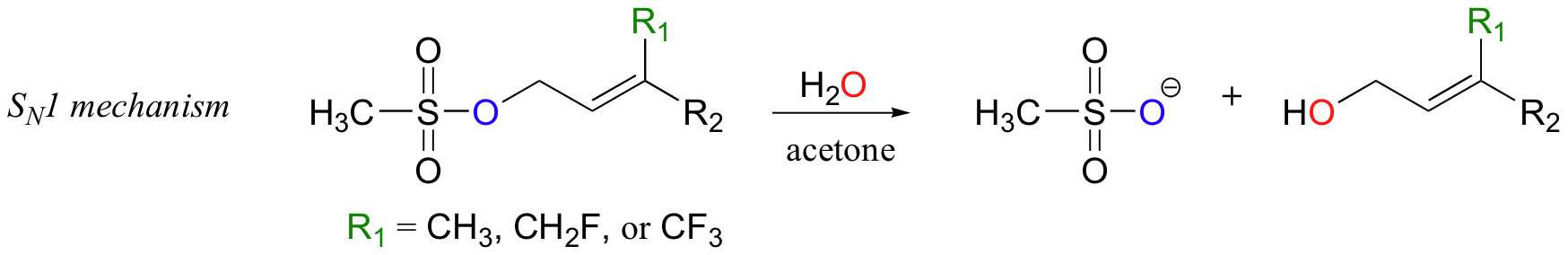
Relative to the compound where R1 = CH3, rates of reaction were 100,000 times slower for the monofluorinated substrate, and 10 million times slower for the trifluorinated substrate (J. Am. Chem. Soc. 1981,103, 3926).
In the model SN2 reaction, however, fluorine substitution on a slightly different substrate resulted in moderately faster rates of substitution (Biochemistry 1977, 16, 5470; J. Am. Chem. Soc. 1962, 84, 3157).
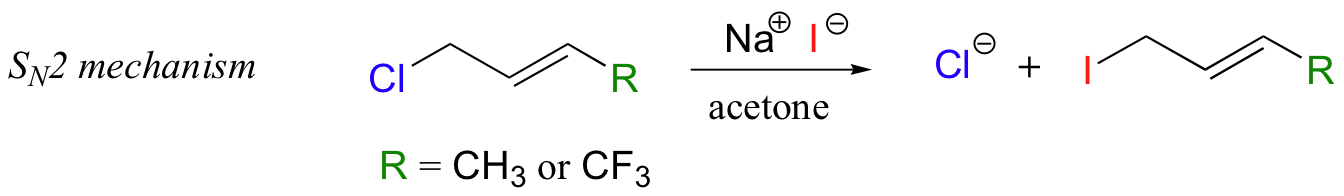
With the protein farnesyltransferase reaction, monofluorination of the substrate resulted in a 10-fold lower reaction rate, while the trifluorinated substrate analog reacted at a rate approximately 1000 times slower than the normal substrate. Clearly, the fluorine substitutions slowed the reaction down, but the observed rate reductions were not nearly as dramatic as those for the model SN1 reaction. The researchers concluded that this reaction mechanism is a hybrid: mainly SN2, but with some SN1 character. In other words, a fully-charged carbocation intermediate does not form, but there is a late transition state: the bond between the electrophilic carbon and the oxygen of the pyrophosphate leaving group has come close to breaking by the time the thiolate approaches.
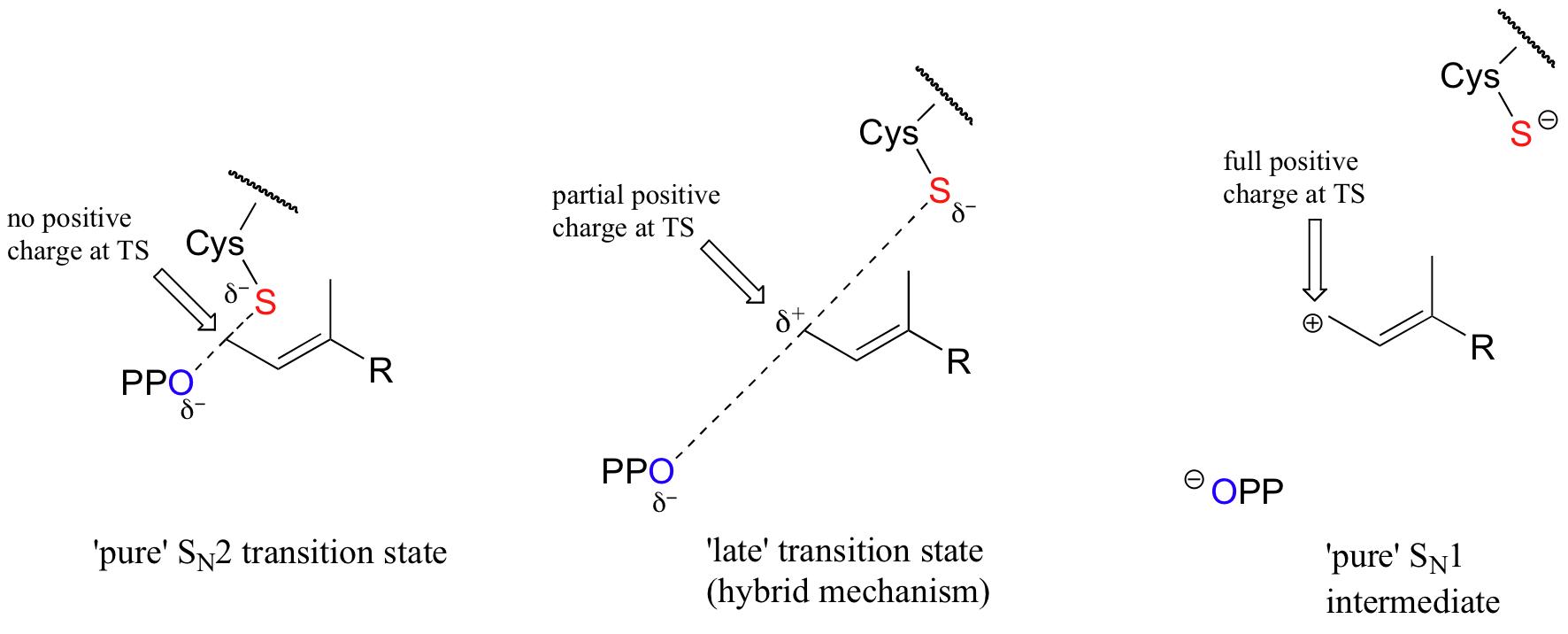
As the reaction proceeds from ground state to transition state, the partial positive charge on the electrophilic carbon increases, which explains the moderately decelerating effect of the electron-withdrawing fluorines.
Exercise 9.3: The enzyme AMP - DMAPP synthase catalyzes the following reaction:
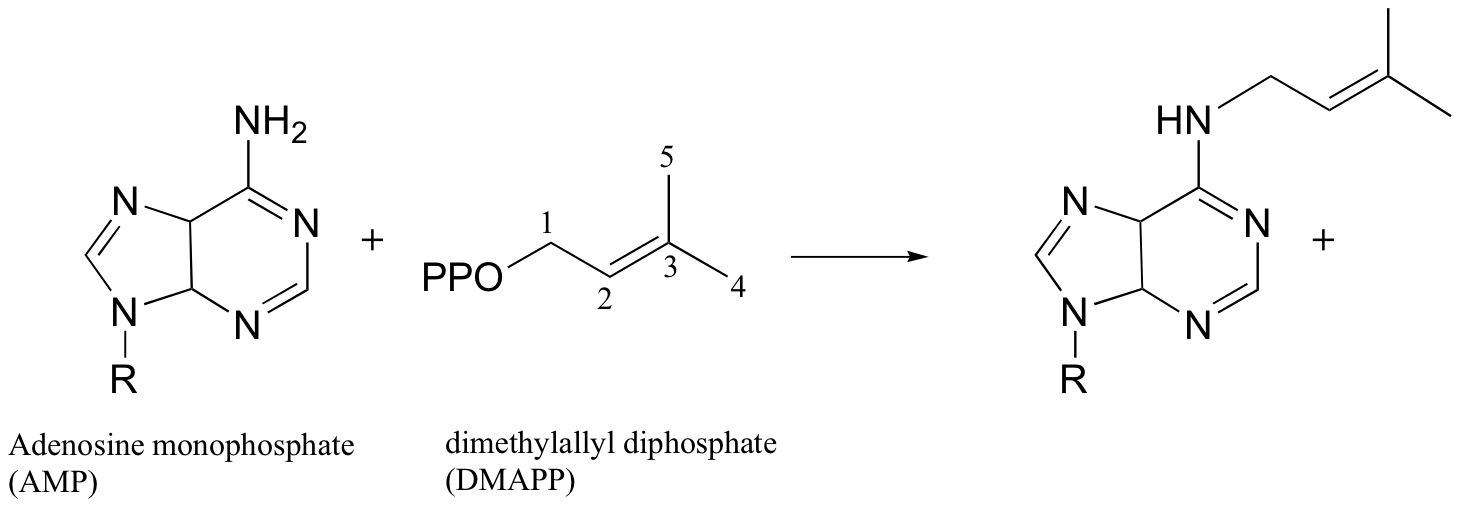
Would you expect fluorine substitution at carbon #5 of the DMAPP substrate to slow the rate of this reaction to a greater or lesser extent than what was observed for the protein farnesyltransferase reaction? Explain your reasoning.
Exercise 9.4: Look again at the two 'model' SN1 and SN2 reactions referred to in the study described in this section. What aspects of each reaction would lead you to predict their mechanisms, if you had not been told?
9.3C: The zinc-thiolate interaction in protein prenyltransferase - 'tuning' the nucleophile
The nucleophile in the substitution reaction catalyzed by protein farnesyltransferase is thought to be a deprotonated thiolate ion (RS-) rather than a thiol. Evidence for a thiolate nucleophile is based on UV spectroscopy experiments (Biochemistry 1999, 38, 13138). Thiolates, unlike thiols, have a characteristic UV absorbance at 236 nm. Absorbance at 236 nm was observed to increase upon binding of the protein substrate to the enzyme, indicating that as the protein substrate enters the enzyme's active site, the critical cysteine is deprotonated. When the substrate protein is free in solution, the pKa of the cysteine thiol is determined experimentally to be approximately 8.5, meaning that it should be mainly protonated at neutral pH. The enzyme, through its interactions with the substrate protein, is able to lower the effective pKa of the cysteine to the point where the thiolate ion is the predominant species. This is accomplished primarily by coordination of the cysteine sulfur atom to a Zn2+ ion that is bound in the active site of the enzyme: the positive charge on the zinc ion balances the negative charge of the cysteine thiolate.
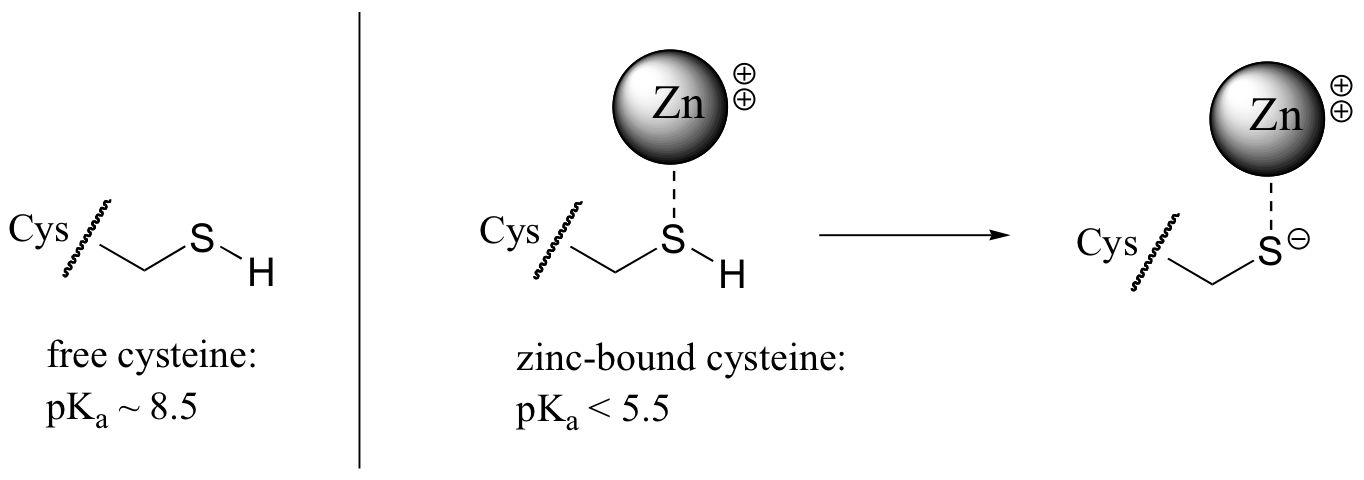
Evidence suggests that coordination to the zinc ion lowers the pKa of the cysteine thiol by approximately 3 pKa units.
This raises an intriguing question that is fundamental to our understanding of how enzymes catalyze reactions. The job of the zinc ion is to lower the pKa of the substrate cysteine, and it does this by stabilizing the negative charge on the conjugate base (thiolate). However, if the zinc stabilizes the thiolate too much, the thiolate will be a less reactive - and thus less effective - nucleophile! If the zinc-thiolate interaction is too strong, in other words, the thiolate will not be able to dissociate from the metal, and nucleophilic attack will not occur. A delicate energetic balance between stability and reactivity is at work here: the zinc must provide just the right amount of charge stabilization to lower the pKa and ensure deprotonation of the thiol, but not so much that the thiolate is no longer reactive enough as a nucleophile.
In effect, the overall positive charge of the zinc ion, and by extension its ability to stabilize the thiolate, must be 'tuned' to achieve the optimal degree of stabilization. How is this accomplished? The answer lies in the way that the zinc ion is specifically bound in the active site of the enzyme. In addition to its interaction with the substrate cysteine, the zinc ion is also coordinated to three amino acid residues from the enzyme: a cysteine, a histidine, and an aspartate.
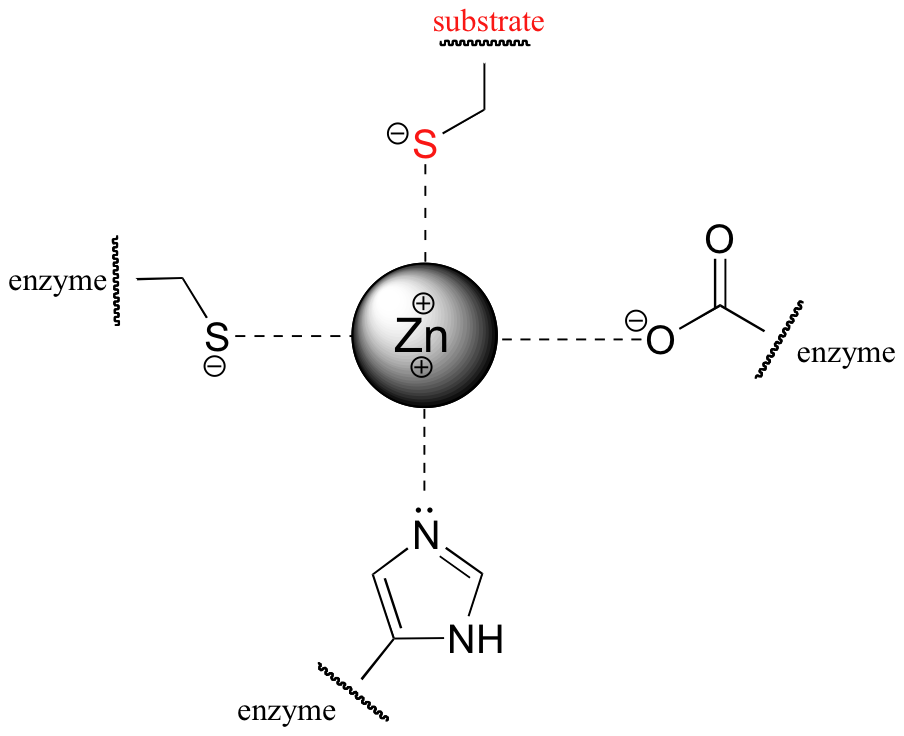
These three ligands, to differing degrees, donate electron density to the zinc, making it less positive. By fine-tuning the overall degree of positive charge on the zinc ion, the enzyme is able to control the extent to which the negative charge on the substrate thiolate is balanced, ensuring that the cysteine pKa is lowered just enough to ensure deprotonation without compromising nucleophilicity.
Researchers studying this phenomenon used site-directed mutagenesis (section 6.5C) to change both of the negatively-charged zinc-coordinating residues (aspartate and cysteine) to neutral histidines, resulting in a zinc center with a substantially greater overall positive charge (Biochemistry 2002, 41, 10554). While this mutant enzyme was able to bind zinc and the two substrates almost as well as the wild-type, no enzymatic activity was detected. It is likely that the substrate cysteine is deprotonated upon binding to the enzyme, but the resulting zinc-thiolate bond is so strong (due to the greater positive charge in the zinc ion) that the thiolate is unable to break away and attack the electrophilic carbon of the farnesyl diphosphate substrate. In effect, the thiolate is 'stuck' on the zinc, and thus is not able to act as a nucleophile.
If you look back at section 8.5C and think about the reasons why synthetic chemists use polar aprotic solvents in laboratory SN2 reactions, you’ll see that many of the same ideas are in play.