
3.4: Constantes de particionamiento y partición
- Page ID
- 70520

3.4. Constantes de particionamiento y partición
3.4.1. Propiedades químicas relevantes
Autores: Joop Hermens, Kees van Gestel
Revisor: Steven Droge, Monika Nendza
Objetivos de aprendizaje
Deberías ser capaz de:
- definir el concepto de hidrofobicidad y explicar qué propiedades químicas afectan a la hidrofobicidad.
- definir qué propiedades de un químico afectan su tendencia a evaporarse del agua.
- calcular fracciones ionizadas para ácidos y bases.
Palabras clave: Hidrofobicidad, coeficientes de partición octanol-agua, volatilidad, constante de la Ley de Henry, químicos ionizados
Introducción
Diferentes procesos afectan el destino de un químico en el medio ambiente. Además de la transferencia e intercambio entre compartimentos (aire-agua-sedimiento/suelo-biota), también la degradación determina la concentración en cada uno de estos compartimentos (Figura 1).
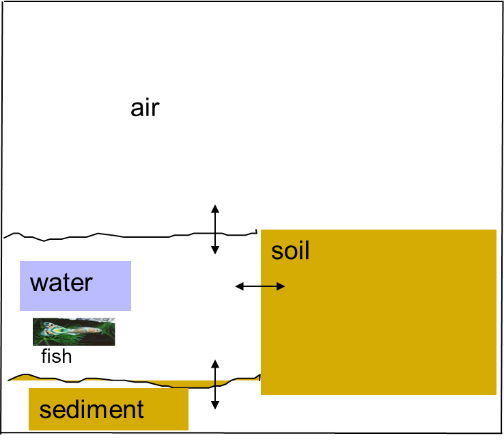
Algunos de estos procesos se discuten en otras secciones (ver secciones sobre Sorción y Degradación ambiental de productos químicos). Algunos químicos se evaporarán fácilmente del agua al aire, mientras que otros permanecen principalmente en la fase acuosa o se sorben para sedimentar y acumularse en la biota.
Estas diferencias están relacionadas con solo unas pocas propiedades básicas:
- Hidrofobicidad (tendencia de una sustancia a escapar de la fase acuosa)
- Volatilidad (tendencia de una sustancia a vaporizarse)
- Grado de ionización
Hidrofobicidad
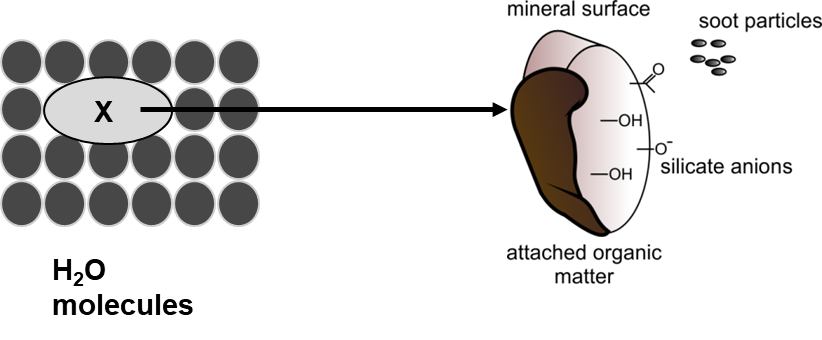
Hidrofobicidad significa miedo (fóbico) al agua (hidro). Un químico hidrófobo prefiere “escapar de la fase acuosa” o en otras palabras “no le gusta disolverse en agua”. Las moléculas de agua están estrechamente unidas entre sí a través de enlaces de hidrógeno. Para que un químico se disuelva en agua, se debe formar una cavidad en la fase acuosa (Figura 2) y esto costará energía.
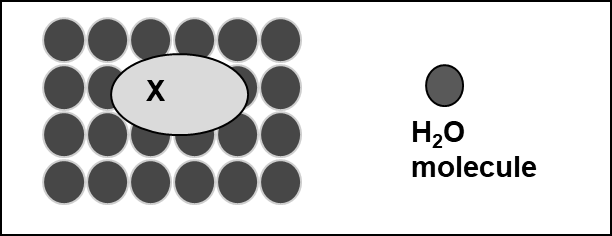
La hidrofobicidad depende principalmente de dos propiedades moleculares:
- Tamaño molecular
- Polaridad/capacidad de interactuar con moléculas de agua, por ejemplo a través de enlaces de hidrógeno
Se necesita más energía para que un químico de mayor tamaño cree la cavidad haciendo que el químico sea más hidrófobo, mientras que las interacciones del químico con el agua favorecerán su disolución haciéndola menos hidrófoba. La Figura 3 muestra productos químicos con hidrofobicidad creciente con aumento de tamaño y una hidrofobicidad decreciente por la presencia de grupos polares (amino o hidroxi).
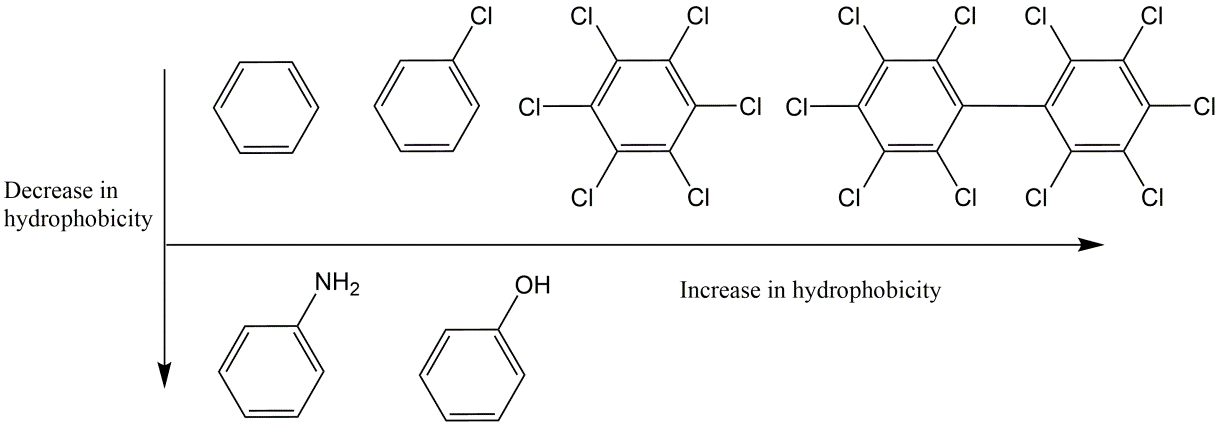
La mayoría de los productos químicos hidrófobos son microcontaminantes orgánicos no polares. Ejemplos bien conocidos son los hidrocarburos clorados, como los bifenilos policlorados (PCB) y los hidrocarburos aromáticos policíclicos (HAP). La solubilidad en agua de estos químicos en general es bastante baja (en el orden de unos pocos ng/L hasta unos pocos mg/L).
La naturaleza hidrofóbica determina principalmente la distribución de estos químicos en el agua y los sedimentos o el suelo y su absorción a través de las membranas celulares. Los átomos adicionales de Cl o Br en una sustancia química, así como unidades adicionales (CH) x, aumentan el tamaño molecular y, por lo tanto, la hidrofobicidad de un químico. El aumento del volumen molecular requiere una cavidad más grande para disolver el químico en el agua, mientras que solo interactúan con las moléculas de agua a través de las interacciones de VanderWaals.
Los grupos polares, como las unidades -OH y -NH en los productos químicos aromáticos de la Figura 3, pueden formar enlaces de hidrógeno con el agua y, por lo tanto, reducir sustancialmente la hidrofobicidad de los productos químicos orgánicos. El enlace de hidrógeno de los sustituyentes hidroxi funciona de dos maneras: El oxígeno de -OH se une a los átomos de H de las moléculas de agua, mientras que el hidrógeno de -OH puede formar puentes a los átomos O de las moléculas de agua. Casi todas las unidades moleculares que consisten en algún tipo de combinación (carbono-oxígeno) reducen la hidrofobicidad de los contaminantes orgánicos, ya que a pesar de que aumentan el volumen molecular interactúan a través de enlaces de hidrógeno (enlaces H) con las moléculas de agua circundantes. Los grupos polares adicionales en una sustancia química suelen disminuir la hidrofobicidad de un químico.
Coeficiente de reparto octanol-agua:
Una simple medida de la hidrofobicidad de los productos químicos, originados en la farmacología, es el coeficiente de reparto octanol-agua, abreviado como K ow (y a veces también llamado P ow o P oct): esta es la relación de concentraciones de un químico en n-octanol y en agua, después establecimiento de un equilibrio entre las dos fases (Figura 4). El grupo -OH en n-octanol permite algunos enlaces de hidrógeno entre las moléculas de octanol en solución, y entre octanol y moléculas disueltas. Sin embargo, la cadena alquílica relativamente larga solo interactúa a través de las interacciones de VanderWaals, y por lo tanto la fuerza de interacción entre las moléculas de octanol es mucho menor que la que existe entre las moléculas de agua, y es energéticamente mucho menos costoso crear una cavidad para disolver cualquier molécula.

Los valores de K ow determinados experimentalmente se utilizaron en la investigación farmacológica para predecir la captación y la actividad biológica de los productos farmacéuticos. Se seleccionó octanol porque parece imitar estrechamente las propiedades moleculares no iónicas de la mayoría de los componentes tisulares, particularmente los fosfolípidos en las membranas. Desde principios de la década de 1970, los valores de K ow también se han utilizado en toxicología ambiental para predecir el peligro y el destino ambiental de los microcontaminantes orgánicos. El octanol también puede imitar parcialmente las propiedades moleculares no iónicas de la mayoría de las fases de materia orgánica que sorben químicos orgánicos neutros en el ambiente biótico y abiótico.
No inesperadamente, la solubilidad en agua se correlaciona negativamente con los coeficientes de partición octanol-agua.
En la práctica, se pueden utilizar tres métodos para determinar o estimar el K ow:
Métodos de equilibrio
En el método del matraz batido (Leo et al., 1971) y el método de 'agitación lenta' (de Bruijn et al., 1989), se determina experimentalmente la distribución de un químico entre octanol y agua. Para los productos químicos altamente lipofílicos (log K ow > 5-6), la solubilidad en agua extremadamente baja, sin embargo, dificulta una determinación analítica confiable de las concentraciones en la fase acuosa. Para tales químicos, estos métodos experimentales no son adecuados. Durante las últimas dos décadas, el uso de columnas generadoras ha permitido cuantificar valores más altos de K ow. Las columnas generadoras son columnas empaquetadas con un material absorbente (por ejemplo, Chromosorb ®) sobre el que se recubre un disolvente hidrófobo apropiado (por ejemplo, octanol) que contiene el compuesto de interés. De esta manera, se crea una gran superficie de interfaz entre las fases lipófila y acuosa, lo que permite un rápido establecimiento de equilibrio. Cuando un gran volumen de agua (saturada de octanol) (típicamente hasta 10 litros) se pasa lentamente a través de la columna, se establece una distribución de equilibrio del compuesto entre el octanol y el agua. El agua que sale de la columna se pasa sobre un cartucho de sorbente sólido para concentrar el compuesto y permitir una cuantificación de la concentración acuosa. De esta manera, es posible determinar de manera más confiable log K ow valores de hasta 6-7.
Cromatografía
Los valores de K ow también pueden derivarse del tiempo de retención en un sistema cromatográfico (Eadsforth, 1986). El uso de cromatografía líquida de alto rendimiento (HPLC) de fase inversa, cromatografía en capa fina o cromatografía de gases da como resultado un factor de capacidad (tiempo de retención relativo; retención del compuesto en relación con una especie química no retenida), que puede usarse para predecir la distribución química sobre octanol y agua. Los sistemas de HPLC han demostrado ser los más exitosos, ya que consisten en fases estacionarias y móviles que son líquidas. Como consecuencia, la naturaleza de las fases se puede organizar más estrechamente para parecerse al sistema octanol-agua. Por supuesto, esto requiere la calibración de los factores de capacidad aplicando el método cromatográfico a una serie de productos químicos con valores bien conocidos de K ow. Los métodos cromatográficos se pueden aplicar de manera confiable para estimaciones de log K ow valores en el rango de 2-8. Para productos químicos más lipófilos, también estos métodos no lograrán predecir de manera confiable los valores de K ow (Schwarzenbach et al., 2003).
Cálculo
También se pueden calcular o predecir valores K ow a partir de parámetros que describen la estructura química de una sustancia química. Varios programas de software están disponibles comercialmente para este propósito, como el programa KOWWIN de la US-EPA. Estos programas hacen uso del llamado método de fragmentos (Leo, 1993; Rekker y Kort, 1979). Este método toma en cuenta la contribución a K ow de diferentes grupos químicos o átomos en una molécula, y además corrige características especiales como impedimento estérico u otras interacciones intramoleculares (ecuación 1):
log K ow = f n + F p (eq.1)
en el que f n cuantifica las contribuciones de cada fragmento n en un producto químico particular (véase por ejemplo el Cuadro 1) y F p representa cualquier interacción intramolecular especial p entre los fragmentos.
Este enfoque de fragmentos ha sido mejorado durante las últimas décadas y está disponible en el programa EPISUITE de la Agencia de Protección Ambiental de Estados Unidos. Otros programas para el cálculo de valores K ow son: ChemProp y ChemAxon de Chemspider.
Cuadro 1. Constantes de fragmentos (K ow) para algunos fragmentos. (del programa EPISUITE)
|
Fragmento |
Constante de fragmento (f) a |
|
-CH 3 carbono alifático |
0.5473 |
|
Carbono Aromático |
0.2940 |
|
-OH hidroxi, unión aromática |
-0.4802 |
|
-N N alifático, una unión aromática |
-0.9170 |
Nota: los cálculos anteriores se dan para los productos químicos no ionizados. La hidrofobicidad de los químicos iónicos también se ve muy afectada por el grado de ionización (ver más abajo).
Los valores K ow también se pueden recuperar de bases de datos como echemportal o ECHA y otras.
Volatilidad
La volatilidad de una sustancia química de la fase acuosa al aire (ver Figura 5) se expresa a través de la constante de la ley de Henry (KH).
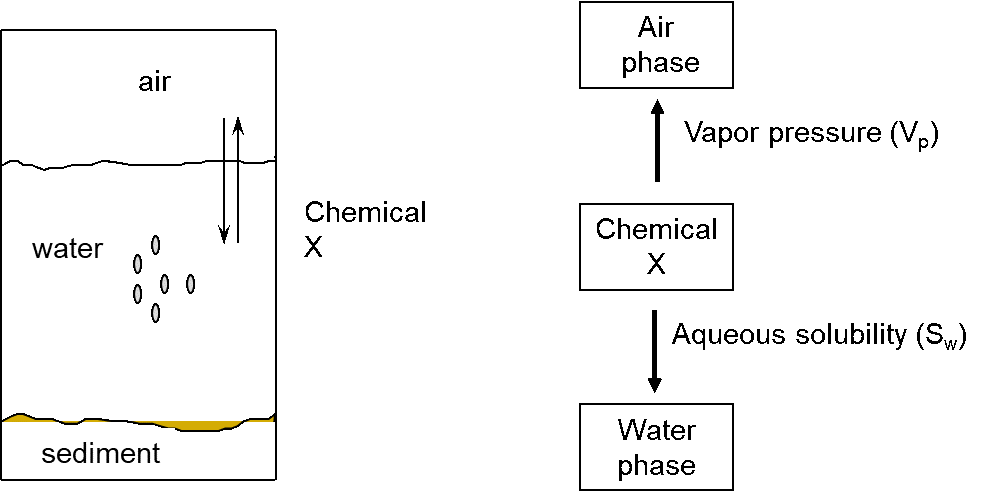
La constante de la ley de Henry (K H, en Pam 3 /mol) es la distribución química entre la fase gaseosa y el agua, como
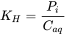 (eq.2)
(eq.2)
donde en un sistema de agua-gas equilibrada:
Caq es la concentración acuosa del químico (unidades en mol/m 3), y P i es la presión parcial del químico en el aire (unidades en Pascal, Pa), que es la presión ejercida por el químico en el volumen total de la fase gaseosa (ocupada por la mezcla de gases la fase gaseosa por encima de la solución acuosa del químico). Tenga en cuenta que Pi es una medida de la concentración en la fase gaseosa, ¡pero aún no en las mismas unidades que la concentración disuelta (discutida más adelante)!
Para compuestos que son ligeramente solubles en agua, K H puede estimarse a partir de:
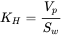 (eq.3)
(eq.3)
donde:
K H: Constante de la ley de Henry (Pam 3/mol), V p es la presión de vapor (saturada) (Pa), que es la presión del químico por encima de la forma condensada pura (líquida) del químico, y S w es la solubilidad máxima en agua (mol/m 3).
La ventaja de la ecuación 3 es que tanto V p como Sw pueden derivarse o estimarse experimentalmente. El fundamento de la ecuación 3 es que dos fuerzas opuestas afectarán la evaporación de un químico del agua al aire:
(i) la presión de vapor (V p) de la sustancia química pura - alta presión de vapor significa más volátil, y
(ii) solubilidad en agua (S w) - alta solubilidad significa menos volátil.
El benceno y el etanol (ver Cuadro 2) son buenas ilustraciones. Ambos químicos tienen una presión de vapor similar, pero la constante de la ley de Henry para el benceno es mucho mayor debido a su solubilidad mucho menor en agua en comparación con el etanol; el benceno es mucho más volátil de una fase acuosa.
Cuadro 2. Coeficientes de partición aire-agua (K aire-agua) calculados para cinco químicos (clasificados por solubilidad acuosa) por la ecuación 3.
|
Químico |
Presión de vapor (Pa) |
Solubilidad (mol/m 3) |
K H (Pa.m 3 /mol) |
K aire-agua (L/L, o m 3 /m 3) |
|
Etanol |
7.5010 3 |
1.2010 4 |
6.2510 -1 |
2.5310 -4 |
|
Fenol |
5.5010 1 |
8.8310 2 |
6.2310 -2 |
2.5210 -5 |
|
Benceno |
1.2710 4 |
2.2810 1 |
5.5710 2 |
2.2510 -1 |
|
Pireno |
6.0010 -4 |
6.5310 -4 |
9.1810 1 |
3.7110 -4 |
|
DDT |
2.0010 -5 |
2.8210 -6 |
7.08 |
2.8610 -3 |
Nota: todos los químicos en equilibrio tienen una concentración mayor (en e.g. mol/L) en la fase acuosa que en la fase gaseosa. De estos cinco, el benceno es el químico más propenso a dejar agua, con una concentración de aire equilibrada aproximadamente 4 veces menor (22.5%) que la concentración disuelta.
Las ecuaciones 2 y 3 se basan en la presión en la fase gaseosa. El destino ambiental a menudo se basa en coeficientes de partición, en este caso el coeficiente de partición aire-agua (K aire-agua). Estos coeficientes de partición son más o menos 'adimensionales', debido a que las concentraciones se basan en volúmenes iguales (como L/L), mientras que K H tiene la unidad Pam 3/mol o algo equivalente a las unidades aplicadas (ecuación 4).
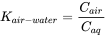 (eq.4)
(eq.4)
donde:
C aire es la concentración en el aire (en por ejemplo mol/m 3) y C ac es la concentración acuosa (en por ejemplo mol/m 3).
K aire-agua se puede calcular a partir de K H de acuerdo con la ecuación 5:
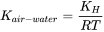 (eq.5)
(eq.5)
donde R es la constante del gas (8.314 m 3 PaK −1 mol −1), y T es la temperatura en Kelvin (Kelvin = o Celsius + 273).
Este uso de “RT” convierte esta concentración de fase gaseosa en una métrica basada en volumen, y aplica la ley de gas ideal que relaciona la presión (P, en Pa) con la temperatura (T, en K), el volumen (V, en m 3) y la cantidad de moléculas de gas (n, en mol), de acuerdo con la constante de gas (R: 8.314 m 3 Pa·K -1 mol -1):
PV = nrt (tenga en cuenta que las unidades de ambos términos se cancelarán) (eq.6)
A 25 o Celcio (298 K), el producto RT es igual a 2477 m 3 PaK -1 mol -1.
En el Cuadro 2 se presentan ejemplos de valores calculados para K aire-agua.
La influencia de la estructura química en la volatilidad de un químico a partir de un solvente depende completamente del costo de crear una cavidad en el solvente (interacciones entre moléculas de solvente) y las interacciones entre el químico y las moléculas de solvente. Para los procesos de partición, la fase gaseosa se considera principalmente como un compartimento inerte sin interacciones químicas (es decir, las moléculas de fase gaseosa casi nunca se tocan entre sí).
Las moléculas de un disolvente fuertemente dipolar como el agua que contienen átomos que pueden interactuar como aceptor de hidrógeno (el O en un grupo OH) y donador de hidrógeno (el H en un grupo OH) interactúan fuertemente entre sí, y cuesta mucha energía crear una cavidad. Este costo aumenta fuertemente con el tamaño molecular, para casi todas las moléculas más que la energía recuperada por las interacciones con las moléculas de disolvente circundantes. Como resultado, para la mayoría de las clases de productos químicos orgánicos, la afinidad con el agua disminuye y la volatilidad del agua al aire aumenta ligeramente con el volumen molecular. Para los productos químicos que no son capaces de reinteractuar a través de enlaces de hidrógeno, por ejemplo, alcanos, la volatilidad general es mucho mayor que para los químicos que tienen interacciones específicas con moléculas de agua además de van der Waals.
Grado de ionización
Los ácidos y bases pueden estar presentes en forma neutra (HA y B) o ionizada (A - y BH +, respectivamente). Para los ácidos, la forma neutra (HA) está en equilibrio con la forma aniónica (A -) y para las bases la forma neutra (B) está en equilibrio con la forma catiónica (BH +). El grado de ionización depende del pH y la constante de disociación ácida (pKa). En el Cuadro 3 se muestran las ecuaciones para calcular la fracción ionizada para ácidos y bases y en el Cuadro 4 se presentan ejemplos de dos ácidos (fenoles).
Cuadro 3. Cálculo de la fracción ionizada para ácidos y bases.
|
Ácidos |
Bases |
|
|
|
|
|
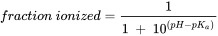 |
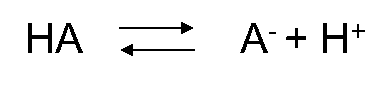
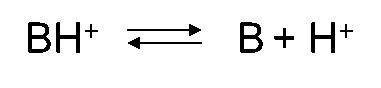
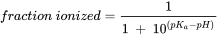
pK a = - log K a, donde K a es constante de disociación de la forma ácida (HA o BH +).
El grado de ionización está así determinado por el pH y el valor de pKa y en otra parte se presentan más ejemplos de varios químicos orgánicos (ver Capítulo Compuestos orgánicos ionogénicos).
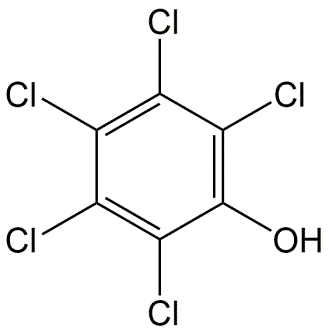
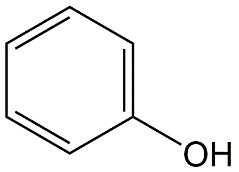
Cuadro 4. El grado de ionización de dos estructuras fenólicas (ácidos).
|
Pentaclorofenol |
Fenol |
|
|
|
|
pKa = 4.60 |
pKa = 9.98 |
|
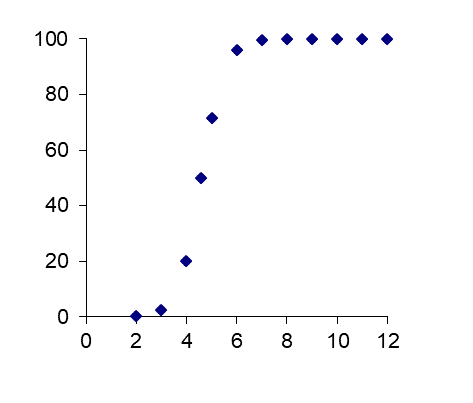
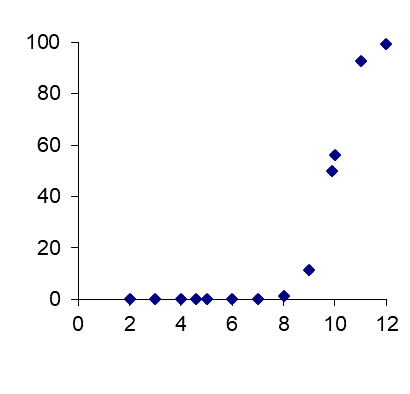
% ionizado versus pH
|
% ionizado versus pH
|
|
a pH de 7.0:99.6% ionizado |
a pH de 7.0:0.1% ionizado |
Ejemplos de varios químicos orgánicos se presentan en otra parte (ver sección sobre Compuestos orgánicos ionogénicos).
El destino de los químicos iónicos es muy diferente al de los químicos no iónicos. El coeficiente de sorción sedimento-agua de las especies aniónicas es sustancialmente (>100x) menor que el de las especies neutras. Si el porcentaje de ionización es menor de ~99% (a un pH 2 unidades por encima del pKa), la sorción del anión puede descuidarse (K d sigue dominada por las especies neutras > 1%) (Schwarzenbach et al., 2003). La razón de la baja afinidad de sorción de la forma de ácido aniónico es doble: los aniones son mucho mejor solubles en agua, pero también la mayoría de las partículas de sedimento (arcilla, materia orgánica, silicatos) están cargadas negativamente y rechazan electrostáticamente el químico cargado de manera similar. En ese caso el coeficiente de sorción K d puede calcularse a partir del coeficiente de sorción de la forma no iónica y la fracción de la forma no ionizada (α):
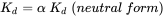 (eq. 7)
(eq. 7)
En ambientes donde el pH es tal que el fraccionamiento ácido neutro2 < 1% (when pH > unidades por encima del pKa), la sorción de las especies aniónicas al suelo/sedimento puede contribuir significativamente al “coeficiente de distribución” general de ambas especies ácidas.
Para los productos químicos ambientales básicos de preocupación, entre los que se encuentran muchas drogas ilícitas (por ejemplo, anfetamina, cocaína) y drogas no ilícitas (por ejemplo, la mayoría de los antidepresivos, betabloqueantes), las formas protonadas están cargadas positivamente. Estos cationes orgánicos también son mucho más solubles en agua que la forma neutra, pero al mismo tiempo son atraídos electrostáticamente por las superficies de sedimentos cargados negativamente. Como resultado, la afinidad de sorción de los cationes orgánicos al sedimento no debe considerarse despreciable en relación con las especies neutras. Los procesos de sorción, sin embargo, pueden diferir fuertemente para las especies de base neutra y las especies de base catiónica. Diversos estudios han demostrado que la afinidad de sorción de especies de base catiónica a DOM o sedimentos es aún más fuerte que la de las especies neutras.
Referencias
De Bruijn, J., Busser, F., Seinen, W., Hermens, J. (1989). Determinación de los coeficientes de reparto octanol/agua para químicos orgánicos hidrófobos con el método de “agitación lenta”. Toxicología y Química Ambiental 8, 499-512.
Eadsforth, C.V. (1986). Aplicación de HPLC de fase inversa para la determinación de coeficientes de partición. Ciencia Pesticida 17, 311-325.
Leo, A., Hansch, C., Elkins, D. (1971). Coeficientes de partición y sus usos. Revisiones Químicas 71, 525-616.
Leo, A.J. (1993). Cálculo de log P (oct) a partir de estructuras. Revisiones Químicas 93, 1281-1306.
Rekker, R.F., de Kort H.M. (1979). La constante fragmentaria hidrófoba; una extensión a un conjunto de 1000 puntos de datos. Eur. J. Med. Chem. - Chim. Tther. 14:479-488.
Schwarzenbach RP, Gschwend PM, Imboden DM (Eds.) 2003. Química orgánica ambiental. Wiley, Nueva York, NY, EUA.
Lectura adicional:
Mackay, D., Boethling, R.S. (Eds.) 2000. Manual de métodos de estimación de propiedades para productos químicos: salud ambiental y ciencias. Prensa CRC.
van Leeuwen, C.J., Vermeire, T.G. (Eds.) 2007. Evaluación de riesgos de productos químicos: Una introducción. Springer, Dordrecht, Países Bajos
Explicar el término hidrofobicidad y mencionar dos propiedades principales que afectan la hidrofobicidad de los productos químicos.
Cuál es la definición de K ow. Clasifique los siguientes productos químicos de la Tabla siguiente de bajo a alto K ow. 1. Pentaclorobenceno, 2. Monoclorobenceno, 3. Monocloroanilina, 4. DDT.
|
1.Pentaclorobenceno |
|
|
2.Monoclorobenceno |
|
|
3.Monocloroanilina |
|
|
4.DDT |
|
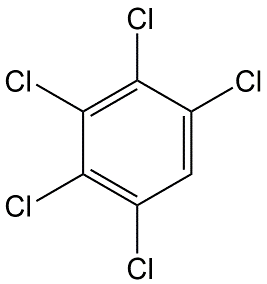
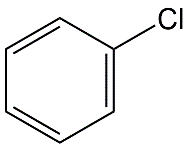
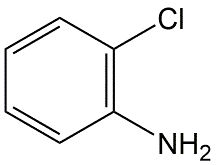
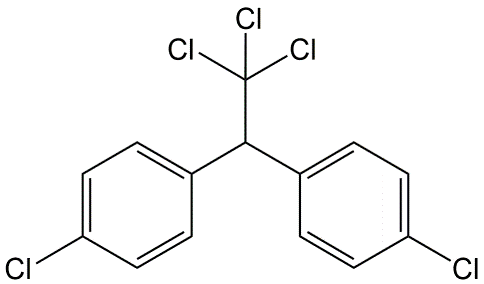
Clasificar los químicos en la Tabla según su volatilidad como compuestos puros y su solubilidad en agua y volatilidad del agua.
Cuales dos propiedades básicas determinan la volatilidad de un químico del agua al aire.
Calcular el porcentaje ionizado para 2,3,4-triclorofenol (pKa = 4.6) a pH 3, 5, 7 y 9.
3.4.2. Sorción
Autores: Joop Hermens
Revisores: Kees van Gestel, Steven Droge, Philipp Mayer
Objetivos de Inclinación:
Deberías ser capaz de:
- comprender por qué la información sobre la sorción es importante para la evaluación de riesgos
- dar ejemplos que ilustran la importancia de la sorción para la evaluación de riesgos
- entender el concepto de isotermas de sorción
- estar familiarizado con diferentes isotermas de sorción (lineal, Freundlich, Langmuir).
Palabras clave: Isoterma de sorción, absorción y adsorción, materia orgánica, modelo Freundlich, modelo Langmuir, contenido de carbono orgánico
Introducción
Los procesos de sorción tienen una influencia importante en el destino de los químicos en el medio ambiente (Recuadro 1). En general, la sorción se define como la unión de un químico disuelto o gaseoso (el sorbato) a una fase sólida (el sorbente) y esto puede implicar diferentes procesos, incluyendo:
(i) unión de sustancias químicas disueltas del agua a sedimentos y suelos
y ii) unión de sustancias químicas en fase gaseosa del aire a suelos, plantas y árboles.
La información sobre la sorción es relevante debido a una serie de razones:
- la sorción controla el destino real y, por lo tanto, el riesgo de (muchos) contaminantes orgánicos e inorgánicos en el medio ambiente,
- los productos químicos absorbidos no pueden evaporarse, no están disponibles para la descomposición (foto) química o microbiana, no se transportan tan fácilmente como los químicos disueltos/en fase vapor y no están disponibles para ser absorbidos por los organismos,
- La sorción también juega un papel importante en las pruebas de toxicidad, afectando las concentraciones de exposición.
|
Caja 1. El Biesbosch es una zona de humedales en los Países Bajos, un área entre los ríos Rin y Mosa y estuarios que están conectados con el Mar del Norte. El flujo de agua es relativamente bajo y como consecuencia hay una fuerte sedimentación de partículas desde el agua hasta el sedimento. Los químicos presentes en el agua se adsorben fuertemente a estas partículas, que en el pasado estaban contaminadas con contaminantes orgánicos hidrófobos como dioxinas y PCB. Las concentraciones de estos compuestos orgánicos en los sedimentos siguen siendo relativamente altas debido a que son altamente persistentes. La razón de esta persistencia de estos compuestos es que estos compuestos sorbidos no están fácilmente disponibles para su degradación por bacterias. Además, las concentraciones en organismos que viven cerca o en el sedimento son altas. Estas concentraciones son tan altas que la pesca de anguila, por ejemplo, no está permitida en la zona. Este ejemplo muestra la importancia de los procesos de sorción sobre el destino, pero también sobre los efectos en el medio ambiente. |
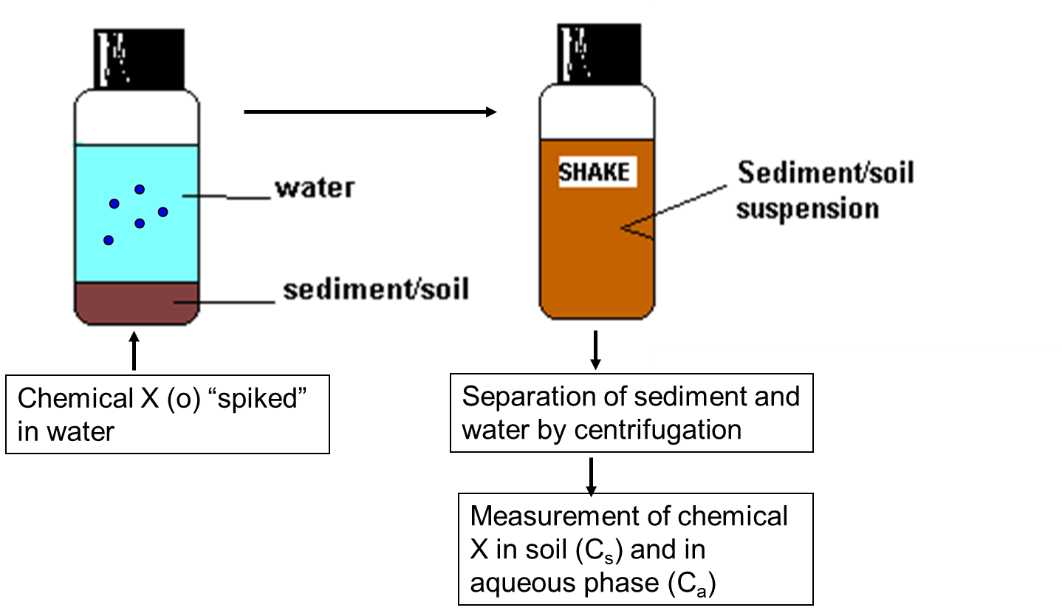
La medición de la sorción es un procedimiento sencillo. Un químico X se agrega (agrega) a la fase acuosa en presencia de una cierta cantidad de la fase sólida (sedimento o suelo). El químico se absorbe a la fase sólida y cuando el sistema está en equilibrio, se miden las concentraciones en el sedimento (C s) y en la fase acuosa (C a). La fase sólida se recoge mediante centrifugación o filtración.
El coeficiente de sorción K p (ecuación 1 y recuadro 2) da información sobre el grado de sorción de un químico al sedimento y se define como:
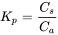 (1)
(1)
|
Caja 2: La concentración de un químico X en el sedimento (C s) es de 30 mg/kg y la concentración en la fase acuosa (C a) es de 0.1 mg/L. El coeficiente de sorción K p = C s/C a = 30 mg/kg/0.1 mg/L = 300 L/kg Anote las unidades de un coeficiente de sorción: L/kg En la evaluación del riesgo ambiental de los productos químicos, es muy útil comprender la fracción de la cantidad total de producto químico (A total) en un sistema que es sorbido (f sorbido) o disuelto (f disuelto) (por ejemplo, debido a un derrame accidental en un río) : f disuelto = A disuelto/A total, y así f sorbido = 1 - f disuelto Esto se relaciona con los coeficientes de sorción de X y el volumen del disolvente y el volumen del material sorbente. La ecuación derivada para calcular f disuelto se basa en el balance de masas del químico A, que relaciona la concentración de X (C) con la cantidad de X (A) en cada volumen (V): C = A/V, y por lo tanto A = C ⋅ V que para un sistema de agua y sedimentos (aire no incluido para simplificación) se refiere a: Un total = A disuelto + A absorbido = C agua ⋅ V agua + C sedimento ⋅ V sedimento = C agua ⋅ V agua + (K p C agua) V sedimento f disuelto = A disuelto/A total = C agua ⋅ V agua/(C agua V agua + K p C agua V sedimento) Esta forma de separar el sedimento C de la ecuación usando K p puede resultar, después de reordenarlo (dividiendo ambas partes de la relación entre C agua ⋅ V agua) a la siguiente ecuación simplificada: f disuelto = 1/(1 + K p ⋅ (V sedimento/V agua)) en esta ecuación, el 'sedimento' puede ser reemplazado por cualquier sorbente, siempre y cuando se utilice el coeficiente de sorción apropiado. Intentemos calcular con X químico desde arriba, en un sedimento húmedo, donde 1L de sedimento húmedo contiene ~ 80% de agua y 20% de sólidos secos. La fracción disuelta de X con K p = 300 Kg/L, es sólo 0.013 en este ejemplo. Así, con 1.3% de X realmente disuelto, esto indica que 98.7% de X se sorbe al sedimento. |
Procesos de sorción
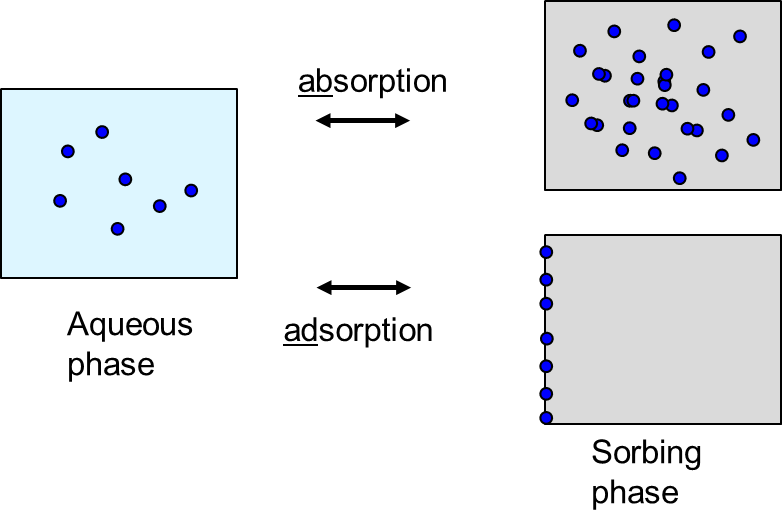
Hay dos procesos principales de sorción (ver Figura 2):
- Absorción - partición (“disolución”) de un químico en una matriz sorbente 3-D. La concentración en la fase de sorción es homogénea.
- Adsorción - unión de un químico a una superficie sorbente 2-D. Debido a que el número de sitios de sorción en una superficie es limitado, los niveles de sorción se reducen a altas concentraciones en la fase acuosa.
Una isoterma de sorción da la relación entre la concentración en un sorbente (sedimento) y la concentración en la fase acuosa y la isoterma es importante para identificar un proceso de sorción.
La absorción de un químico es similar a su partición entre dos fases y comparable a su partición entre dos disolventes. La distribución de un producto químico entre octanol y agua es un ejemplo bien conocido de un proceso de partición (ver Sección 3.4.1 sobre Propiedades químicas relevantes para obtener información más detallada sobre la partición octanol-agua). La isoterma para un proceso de absorción es lineal (Figura 3A) y la pendiente de la gráfica y-x es el coeficiente de sorción K p.
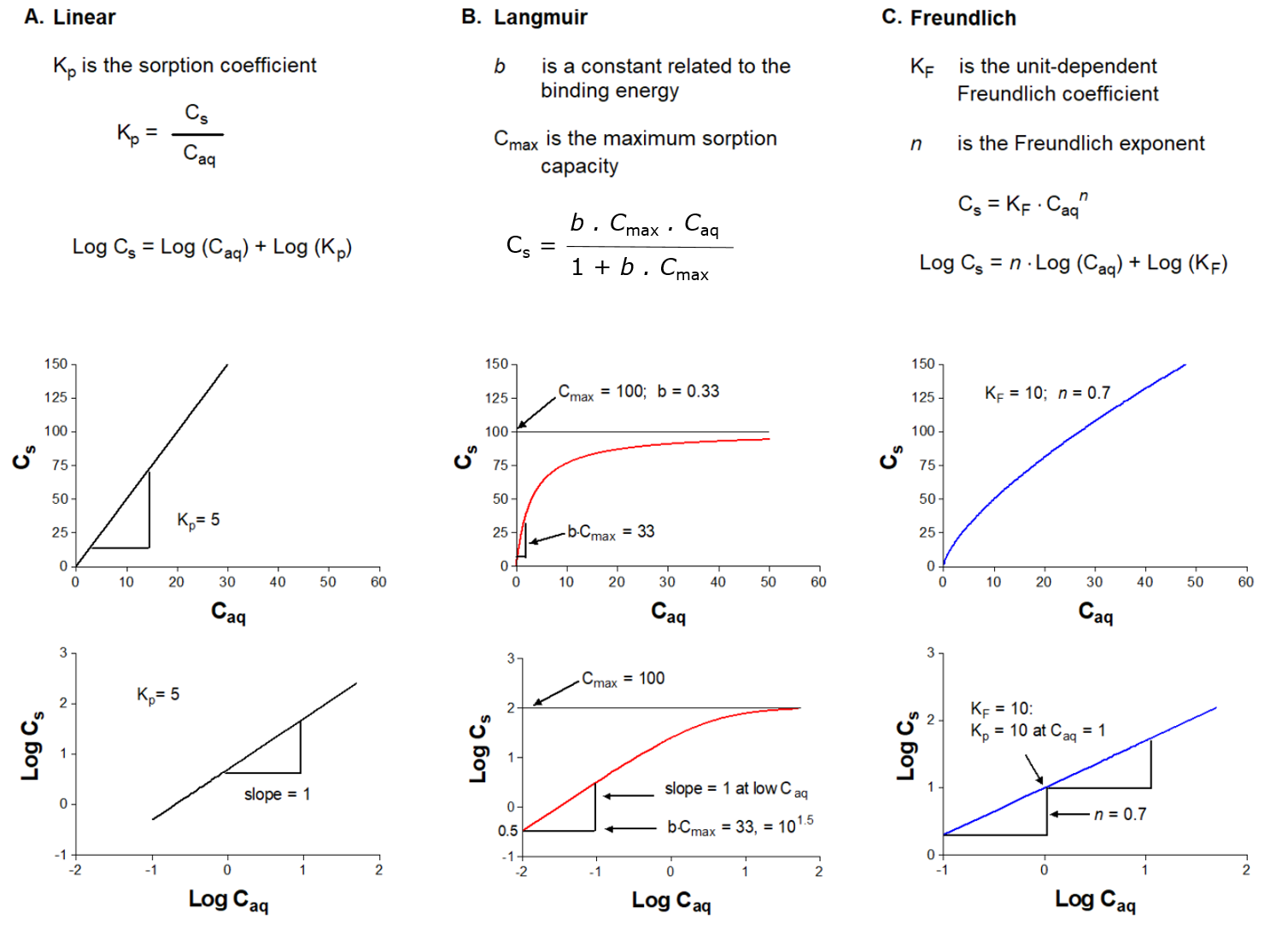
En un proceso de adsorción, donde la fase de sorción es una superficie con un número limitado de sitios de sorción, la isoterma de sorción es no lineal y puede alcanzar una concentración máxima que se adsorbe cuando todos los sitios están ocupados. Un modelo mecanicista para la adsorción es el modelo de Langmuir. Este modelo describe la adsorción de moléculas a superficies homogéneas con energías de adsorción iguales, representadas por el término de energía del sitio de adsorción (b) y un número limitado de sitios de sorción (C max) que pueden saturarse (Figura 3B). El coeficiente de adsorción de Langmuir (K ad) es igual al producto (b ⋅ C max) a concentraciones acuosas relativamente bajas, donde el producto (b ⋅ C aq) << 1 (nótese que el término denominador será entonces ~1). De hecho, la curva de isoterma en una gráfica a doble escala logarítmica muestra una pendiente de 1 a concentraciones tan bajas, lo que indica linealidad.
Otro enfoque matemático para describir la sorción no lineal es la isoterma de Freundlich (Figura 3C), donde KF es la constante de sorción de Freundlich y n es el exponente de Freundlich que describe la no linealidad del proceso de sorción. Usando valores logarítmicos para concentraciones acuosas y sorbidas, la isoterma de Freundlich se puede reescribir como:
Log C s = n ⋅ log C aq + log K F (eq. 2)
Esto convenientemente produce una relación lineal (así como y = ax + b) entre log Cs y log Caq, con una pendiente igual a n y la abscisa (punto de cruce con el eje Y) igual al log KF. Esto permite un ajuste fácil de líneas de tendencia lineal a través de conjuntos de datos experimentales. Cuando n = 1, la isoterma es lineal, y equivale a la de absorción. En caso de saturación de los sitios de sorción en la fase sólida, 1/n será menor que 1. Sin embargo, la isoterma de Freundlich también puede producir un valor 1/n > 1; esto puede ocurrir, por ejemplo, si el producto químico que se sorbe forma una capa que sirve como nueva fase de sorción y se describen ejemplos para tensioactivos.
Fases de sorción
Los suelos y sedimentos pueden mostrar grandes variaciones en la composición y distribución del tamaño de partícula. Los principales componentes de suelos y sedimentos son:
|
Arena |
63 - 2 mm |
|
Limo |
2 - 63 µm |
|
Arcilla |
<2 µm |
|
Materia orgánica |
incluye, por ejemplo, detritus, ácidos húmicos, especialmente asociados con las fracciones de arcilla y limo |
|
CaCo 3 |
La Figura 4 da una imagen esquemática de un sedimento o partícula de suelo. Además de la presencia de minerales arcillosos y (suelo o sedimento) materia orgánica (SOM), el sedimento y el suelo pueden contener partículas de hollín (un residuo de combustión).
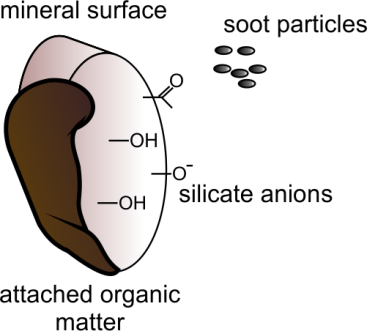
La materia orgánica se forma tras la descomposición de material vegetal y tejidos animales o microbianos muertos. Al descomponerse el material vegetal, los primeros grupos orgánicos que se liberan son los ácidos fenólicos, algunos de los cuales tienen una alta afinidad por la complejación de metales. Un ejemplo es el ácido salicílico (ácido o-hidroxibenzoico), que se presenta en altas concentraciones en hojas de sauces, álamo y otros árboles caducifolios. Una mayor descomposición del material vegetal puede resultar en la formación de ácidos húmicos, ácidos fúlvicos y humina. Los ácidos húmico y fúlvico contienen una serie de grupos funcionales, como los grupos carboxilo- (COOH), carbonilo- (=C=O), hidroxilo fenólico- (-OH), metoxi- (-OCH3), amino- (-NH2), imino (=NH) y sulfhidrilo (-SH) (ver para más detalles la sección sobre Suelo).
Los productos químicos orgánicos hidrófobos se adsorben principalmente a materia orgánica. Debido a que la materia orgánica tiene las características de un disolvente, la sorción es claramente un proceso de absorción y la isoterma de sorción es lineal. Debido a que la unión es principalmente a la materia orgánica, el coeficiente de sorción (K p) depende de la fracción de materia orgánica (f om) o de la fracción de carbono orgánico (f oc) presente en el suelo o sedimento. Tenga en cuenta que como regla general, la materia orgánica contiene 58% de carbono orgánico (f oc = 0.58f om). La Figura 5A muestra el incremento del coeficiente de sorción al aumentar la fracción de carbono orgánico en suelos y sedimentos. Para llegar a un parámetro más intrínseco, los coeficientes de sorción suelen normalizarse a la fracción materia orgánica (K om) o carbono orgánico (K oc). Estos valores K oc o K om son menos dependientes del tipo de sedimento o suelo (Figura 5B).
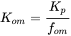 (3)
(3)
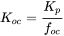 (4)
(4)
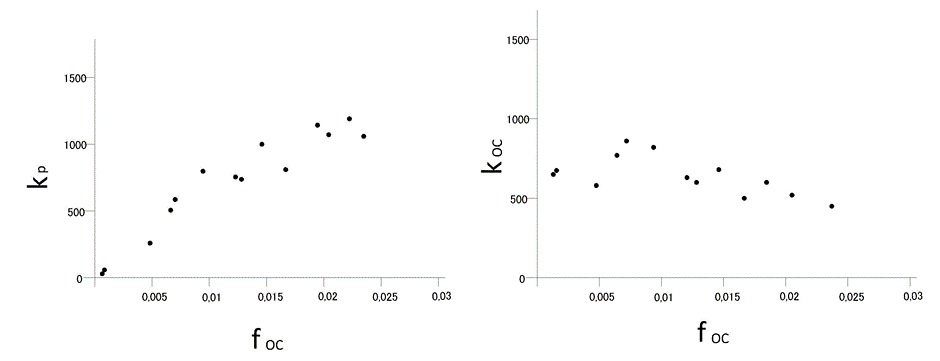
Los químicos hidrófobos pueden tener una afinidad muy alta por las partículas de hollín en relación con la afinidad por SOM. Si un sedimento contiene hollín, los valores de K p suelen ser más altos de lo previsto en función de la fracción de carbono orgánico en la materia orgánica (Jonker y Koelmans, 2002).
Referencias
Schwarzenbach, R.P., Gschwend, P.M., Imboden, D.M. (2003). Química Orgánica Ambiental. Wiley, Nueva York, NY, EUA.
Medios, J.C., Wood, S.G., Hassett, J.J., Banwart, W.L. (1980). Sorción de hidrocarburos aromáticos polinucleares por sedimentos y suelos. Ciencia y Tecnología Ambiental 14, 1524-1528.
Jonker, M.T.O., Koelmans, A.A. (2002). Sorción de hidrocarburos aromáticos policíclicos y bifenilos policlorados a hollín y materiales similares al hollín en el ambiente acuoso consideraciones mecanicistas. Ciencia y Tecnología Ambiental 36, 3725-3734.
Lectura sugerida
van Leeuwen, C.J., Vermeire, T.G. (Eds.) (2007). Evaluación de Riesgos de Químicos: Una Introducción. Springer, Dordrecht, Países Bajos. Capítulo 3 y 9.
Schwarzenbach, R.P., Gschwend, P.M., Imboden, D.M. (2003). Química Orgánica Ambiental. Wiley, Nueva York, NY, Estados Unidos. capítulos 9, 11. Información detallada sobre procesos de sorción y mecanismos de sorción.
¿Qué es una isoterma de sorción y cuál es la diferencia entre absorción y adsorción?
¿Por qué la isoterma de sorción a la arcilla no es lineal?
¿Cuáles son las principales fases de sorción en sedimentos o suelos?
3.4.3. QSPRs
Autor: Joop Hermens
Revisor: Steven Droge, Monika Nendza
Objetivos de aprendizaje:
Deberías ser capaz de:
- indicar qué propiedades de los productos químicos se aplican en un QSPR
- enumerar diferentes técnicas para derivar un QSPR
- indican qué interacciones pueden ocurrir entre moléculas.
Palabras clave: Relaciones cuantitativas estructura-propiedad (QSPR), relaciones cuantitativas estructura-actividad (QSAR), coeficientes de partición octanol-agua, enlaces de hidrógeno, técnicas multivariadas.
Introducción
La evaluación de riesgos necesita datos de entrada para parámetros de destino y efecto. Estos datos no están disponibles para muchos de los productos químicos existentes y las predicciones a través de modelos de estimación proporcionarán una buena alternativa a las pruebas reales. Ejemplos de modelos de estimación son Relaciones Cuantitativas Estructura-Propiedad (QSPR) y Relaciones Cuantitativas Estructura-Actividad (QSARs). El término “actividad” se usa a menudo en relación con los modelos de toxicidad, mientras que “propiedad” generalmente se refiere a propiedades físico-químicas o parámetros de destino.
En un QSAR o QSPR, cierto parámetro ambiental está relacionado con una propiedad físico-química o estructural, o una combinación de propiedades.
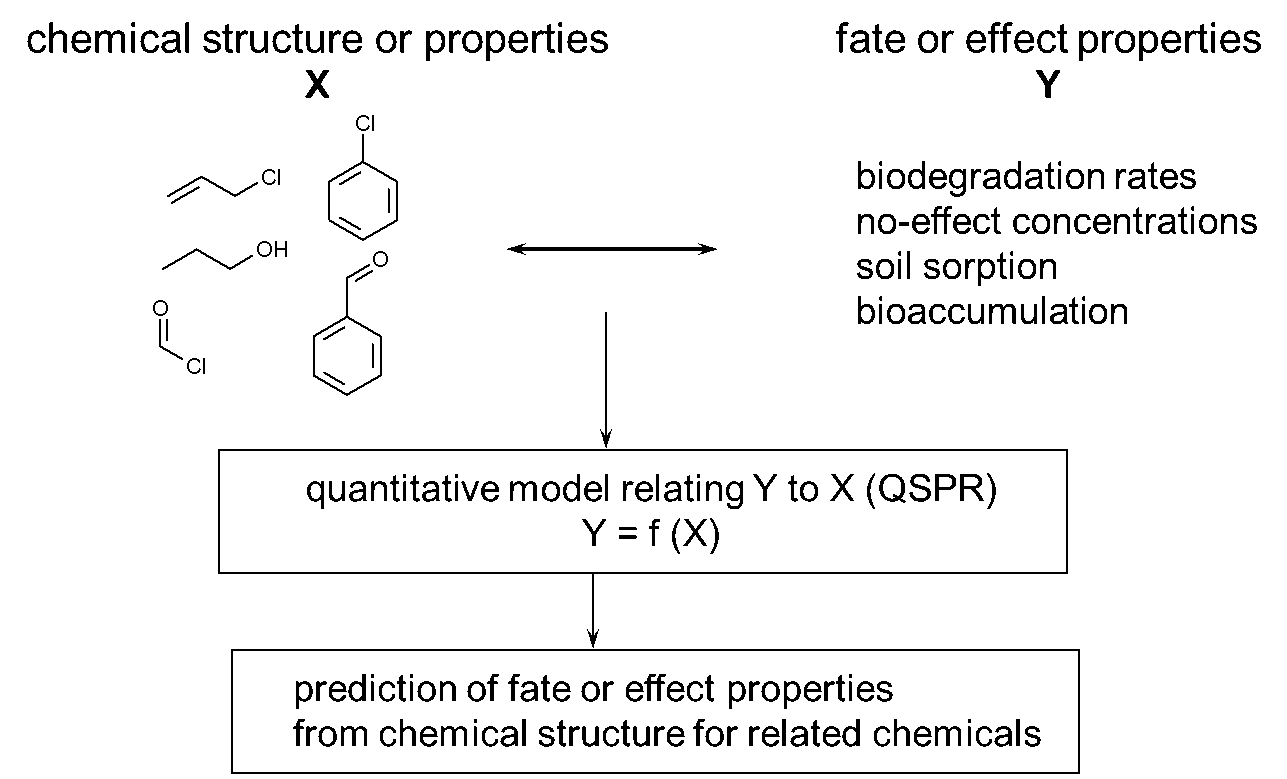
Los elementos en un QSPR o QSAR se muestran en la Figura 1 e incluyen:
- el parámetro para el que se ha desarrollado el modelo de estimación: la variable Y (superior derecha),
- las propiedades del parámetro químico o químico: la variable X (superior izquierda),
- el modelo en sí (centro), y
- la predicción de un parámetro de destino o efecto a partir de las propiedades químicas (abajo).
La variable Y
Se han desarrollado modelos de estimación para muchos criterios de valoración tales como sorción a sedimentos, ácidos húmicos, lípidos y proteínas, degradación química, biodegradación, bioconcentración y efectos ecotóxicos.
La variable X
En el Cuadro 1 se presenta una visión general de los parámetros químicos (la variable X) utilizados en los modelos de estimación. Las propiedades químicas se dividen en tres categorías: (i) parámetros relacionados con la hidrofobicidad, (ii) parámetros relacionados con la carga y distribución de carga en una molécula y (iii) parámetros relacionados con el tamaño o volumen de una molécula. La hidrofobicidad se discute con más detalle en la sección Propiedades químicas relevantes.
Otros enfoques QSPR utilizan gran cantidad de parámetros derivados de gráficos químicos. El software CODESSA Pro, por ejemplo, genera descriptores moleculares (494) y de fragmentos (944), clasificados como (i) constitucionales, (ii) topológicos, (iii) geométricos, (iv) relacionados con la carga y (v) química cuántica (Katritzky et al. 2009). Algunos modelos se basan en fragmentos estructurales en una molécula. Las relaciones lineales de energía libre poliparamétricas (PP-Lfer) utilizan parámetros que representan interacciones entre moléculas (ver bajo PP-Lfer).
Cuadro 1. Ejemplos de parámetros relacionados con hidrofobicidad y parámetros electrónicos y estéricos (la variable X).
|
Parámetros hidrofóbicos |
|
Solubilidad acuosa |
|
Coeficiente de partición octanol-agua (K ow) |
|
Constante de fragmento hidrófobo π |
|
Parámetros electrónicos |
|
Cargas atómicas (q) |
|
Momento dipolo |
|
Acidez del enlace de hidrógeno (donador de enlaces H) |
|
Basicidad del enlace de hidrógeno (aceptor de enlace H) |
|
Constante de Hammett σ |
|
Parámetros estéricos |
|
Superficie Total (TSA) |
|
Volumen Molecular Total (TMV) |
|
Constante de Taft para efectos estéricos (Es) |
El modelo
La mayoría de los modelos se basan en correlaciones entre Y y X. Dicha relación se deriva para un "conjunto de entrenamiento" que consiste en un número limitado de sustancias químicas cuidadosamente seleccionadas. La validez de dicho modelo debe probarse aplicándolo a un "conjunto de validación “, es decir, un conjunto de compuestos para los que se puedan comparar datos experimentales con las predicciones. Se pueden utilizar diferentes técnicas para desarrollar un modelo empírico, tales como:
- presentaciones gráficas,
- ecuaciones lineales o no lineales entre Y y X,
- ecuaciones lineales o no lineales basadas en diferentes propiedades (Y versus X 1, X 2, etc.),
- técnicas multivariadas como Análisis de Componentes Principales (PCA) y Análisis Parcial de Mínimos Cuadrados (PLS).
Las ecuaciones lineales toman la forma:
Y (i) = a 1 X 1 (i) + a 2 X 2 (i) + a 3 X 3 (i) +... + b (1)
donde Y (i) es el valor del parámetro dependiente de la sustancia química i (por ejemplo coeficientes de sorción); X 1 -X 3 (i) son valores para los parámetros independientes (las propiedades químicas) del químico i; a 1 - a 3 son coeficientes de regresión ( generalmente se dan límites de confianza del 95%); b es la intercepción de la ecuación lineal. La calidad de la ecuación se presenta a través del coeficiente de correlación (r) y el error estándar de estimación (es). Cuanto más cerca está r a 1.0, mejor es el ajuste de la relación. Se puede encontrar más información sobre la calidad estadística de los modelos bajo “limitación de QSPR”.
El enfoque clásico en los estudios QSAR y QSPR es el enfoque de Hansch que se desarrolló en la década de 1960. La ecuación de Hansch (Hansch et al., 1963) describe la influencia de los sustituyentes sobre la actividad biológica en una serie de compuestos parentales con un cierto sustituyente (ecuación 2). Los sustituyentes son, por ejemplo, un cierto átomo o grupo químico (Cl, F, B,. OH, NH 2) unido a una estructura de anillo aromático parental.
log 1/C = c π + c' σ + c” E s + c"' (2)
en el que:
C es la concentración molar de una sustancia química con un efecto particular,
n es una constante sustituyente para efectos hidrófobos,
σ es una constante sustituyente para efectos electrónicos, y
E s es una constante sustituyente para efectos estéricos.
c son constantes que se obtienen ajustando datos experimentales
Por ejemplo, la constante de sustituyente hidrófobo se basa en Kow y se define como se define como:
π (X) = log K ow (RX) - log K ow (RH) (3)
donde RX y RH son el compuesto parental sustituido y no sustituido, respectivamente.
Las constantes Hammett y Taft se derivan de manera similar.
Las técnicas multivariadas pueden ser muy útiles para desarrollar relaciones estructura-actividad, en particular en los casos en los que interviene un gran número de parámetros químicos. El Análisis de Componentes Principales (PCA) se puede aplicar para reducir el número de variables en algunos componentes principales. El siguiente paso es encontrar una relación entre Y y X mediante, por ejemplo, análisis de mínimos cuadrados parciales (PLS). La ventaja de PCA y PLS es que puede tratar con un gran número de descriptores químicos y que también puede hacer frente a propiedades colineales (correlacionadas). Más información sobre estas técnicas multivariadas y ejemplos en el campo de la ciencia ambiental son dados por Eriksson et al. (1995).
Relación lineal de energía libre poli-parámetro (PP-Lfer)
El enfoque de PP-Lfer tiene una fuerte base mecanicista porque incluye los diferentes tipos de interacciones entre moléculas (Goss y Schwarzenbach, 2001). Por ejemplo, el coeficiente de sorción de un químico de una fase acuosa al suelo o a los fosfolípidos (el sorbente) depende de la interacción de un químico con el agua y la interacción con la fase sorbente. Una de las fuerzas impulsoras detrás de la sorción es la hidrofobicidad. Hidrofobicidad significa miedo (fobia) por el agua (hidro). Un químico hidrófobo prefiere “escapar de la fase acuosa” o en otras palabras “no le gusta disolverse en agua”. Las moléculas de agua están estrechamente unidas entre sí a través de enlaces de hidrógeno. Para que un químico se disuelva, se debe formar una cavidad en la fase acuosa (Figura 2) y esto costará energía. Los compuestos más hidrófobos suelen tener una sorción más fuerte (ver más información en la sección Propiedades químicas relevantes).
La hidrofobicidad depende principalmente de dos propiedades moleculares:
- Tamaño molecular
- Polaridad/capacidad de interactuar con moléculas de agua, por ejemplo a través de enlaces de hidrógeno
En la interacción con el sorbente (suelo, lípidos de membrana, lípidos de almacenamiento, ácidos húmicos), las interacciones principales son las interacciones de van der Waals y los enlaces de hidrógeno (Cuadro 2). Las interacciones de Van der Waals son atractivas y ocurren entre todo tipo de moléculas y la fuerza depende del área de contacto. Por lo tanto, la fuerza de las interacciones de van der Waals está relacionada con el tamaño de una molécula. Un enlace de hidrógeno es una atracción electrostática entre un hidrógeno (H) y otro átomo electronegativo que lleva un par solitario de electrones. El átomo de hidrógeno suele estar unido covalentemente a un átomo más electronegativo (N, O, F). En el Cuadro 2 se enumeran las interacciones con ejemplos de estructuras químicas.
Un PP-Lfer es una ecuación lineal desarrollada para modelar coeficientes de partición o sorción (K) usando parámetros que representan las interacciones (Abraham, 1993). La ecuación del modelo se basa en cinco descriptores:
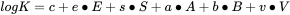 (2)
(2)
con:
|
E |
exceso de refracción molar |
|
S |
parámetro de dipolaridad/polarizabilidad |
|
A |
Acidez del enlace H del soluto (donante del enlace H) |
|
B |
Basicidad del enlace H del soluto (aceptor del enlace H) |
|
V |
volumen molar |
El coeficiente de partición o sorción K puede expresarse como la suma de cinco términos de interacción, describiendo los parámetros en mayúscula las propiedades específicas del compuesto. E depende de la estructura electrónica de valencia, S representa polaridad y polarizabilidad, A es la fuerza donadora del enlace de hidrógeno (HB) (acidez HB), B la fuerza aceptora HB (basicidad HB), V es el llamado volumen característico relacionado con el tamaño de la molécula, y c es una constante. Los parámetros en minúscula expresan las propiedades correspondientes del sistema bifásico respectivo y, por lo tanto, pueden tomarse como la importancia relativa de las propiedades del compuesto para el proceso particular de partición o sorción. En esta sección introductoria, solo nos enfocamos en el factor de volumen (V) y los dos parámetros de enlace de hidrógeno (A y B).
Se han desarrollado numerosos PP-Lfers para todo tipo de procesos ambientales y Endo y Goss (2014) dan una visión general.
Cuadro 2. Tipos de interacciones entre las moléculas y la fase a la que se sorben con ejemplos de químicos (Goss y Schwarzenbach, 2003).
|
Compuesto a) |
Interacciones |
Ejemplos |
|
Apolar |
solo van der Waals |
alcanos, clorobencenos, PCB |
|
Monopolar |
van der Waals + Aceptador H (e-donador) |
alquenos, alquinos, compuestos alquilaromáticos éteres, cetonas, ésteres, aldehídos |
|
Monopolar |
van der Waals + Donante H (aceptor electrónico) |
CHCl 3, CH 2 Cl 2 |
|
Bipolar |
van der Waals + Donante H + aceptor H |
R-NH 2, R2-NH, R-COOH, R-OH |
a) Apolar: ningún grupo polar presente; mono/dipolar: uno o dos grupos polares presentes en una molécula
Ejemplos de QSPR para bioconcentración en peces
Modelo basado en K ow
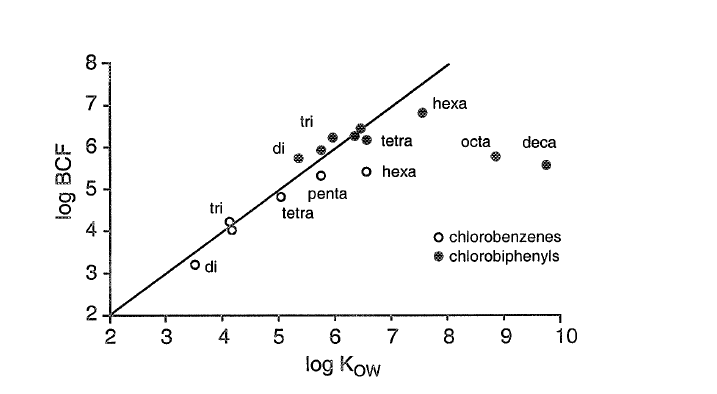
Los modelos predictivos de bioconcentración tienen una larga historia. El coeficiente de partición octanol-agua (K OW) es una buena medida de hidrofobicidad y los factores de bioconcentración (BCF) a menudo se correlacionan con K ow (ver más información en la sección Bioacumulación). El éxito de estos modelos basados en K OW se explicó por la semejanza de la partición en octanol y lípidos a granel en los organismos, al menos para compuestos hidrófobos neutros. Un ejemplo bien conocido de un modelo QSAR lineal para el log BCF (variable Y) basado en el log K OW (variable X) (Veith et al., 1979):
log BCF = 0.85 log K OW - 0.70 (5)
La Figura 3 da un ejemplo clásico de tal correlación para BCF a guppy de una serie de bencenos clorados y bifenilos policlorados. Cuando los químicos lipofílicos son metabolizados, la relación mostrada en la Figura 3 ya no es válida y el BCF será menor de lo previsto basado en K OW. Otra desviación de esta relación BCF-K ow se puede encontrar para químicos altamente lipofílicos con log K ow >7. Para tales químicos, el BCF suele disminuir nuevamente al aumentar el Kow (ver Figura 3). La curva aparente de BCF con Kow como variable X tiende a seguir una curva no lineal con un óptimo en log K ow 7-8. Este fenómeno puede explicarse a partir del tamaño molecular: las moléculas de productos químicos como el decaclorobifenilo pueden ser tan grandes que tienen dificultades para pasar las membranas. Una explicación más probable, sin embargo, es que para los químicos altamente lipofílicos las concentraciones acuosas pueden ser sobreestimadas. No es fácil separar los productos químicos unidos a partículas de la fase acuosa (ver recuadro 1 en la sección sobre Sorción) y esto puede llevar a concentraciones medidas que son superiores a la concentración biodisponible (libremente disuelta) (Jonker y van der Heijden 2007; Kraaij et al. 2003). Por ejemplo, a una concentración de carbono orgánico disuelto (DOC) de 1 mg-DOC/l, un químico con un log Koc de 7 estará 90% unido a partículas, y esta fracción unida no forma parte de la concentración disuelta que se equilibra con el tejido (pescado). Esto demuestra que estos modelos también son interesantes porque pueden mostrar tendencias en los datos que pueden conducir a una mejor comprensión de los procesos.
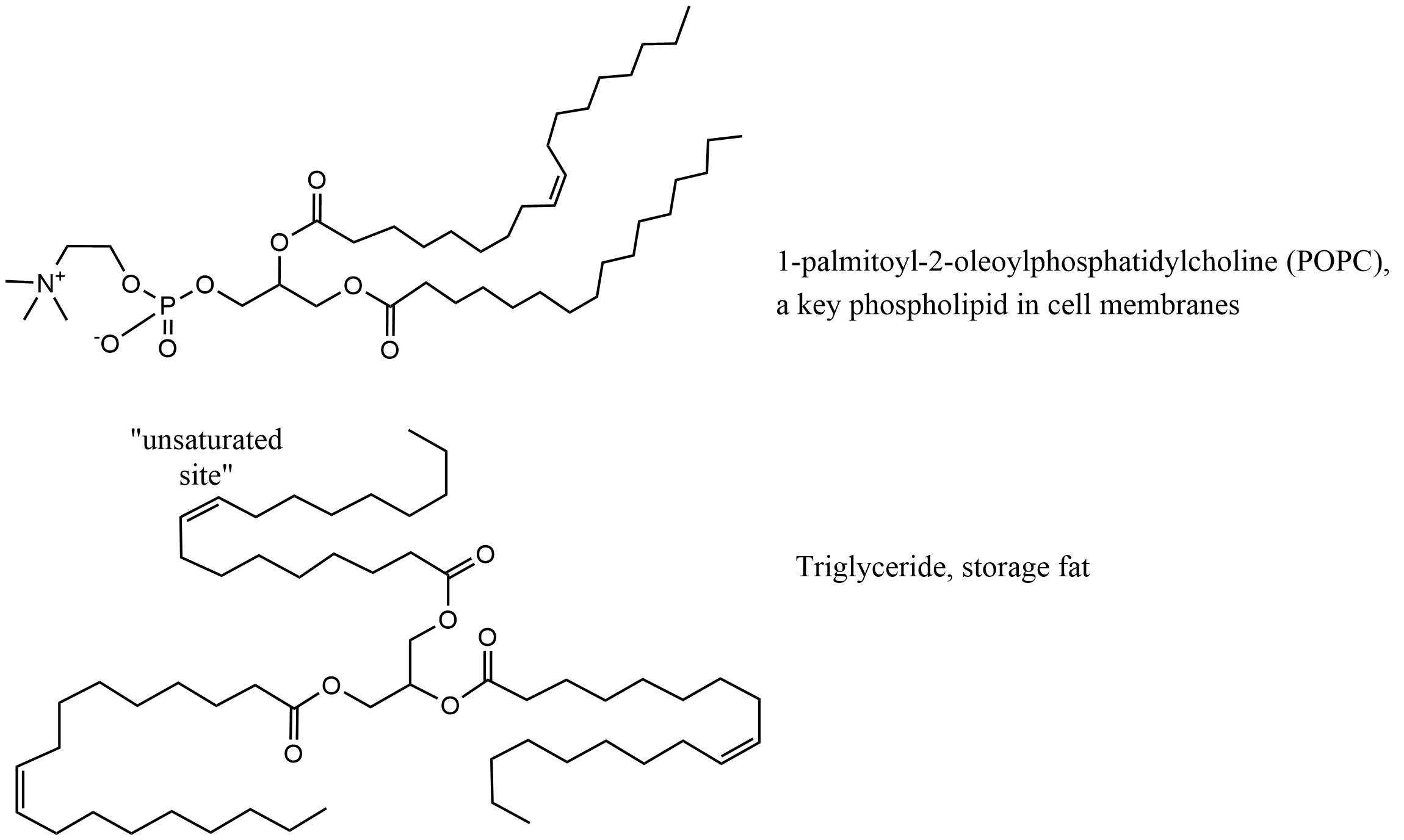
Ejemplos de QSPR para sorción a lípidos
Los modelos basados en K ow son exitosos porque el octanol probablemente tiene propiedades similares a las de los lípidos de los peces. Existen varios tipos de lípidos y los lípidos de membrana tienen diferentes propiedades y estructura que por ejemplo los lípidos de almacenamiento (ver Figura 4, y más detalles en la sección sobre Biota). Los modelos de BCF más refinados incluyen la separación de lípidos de almacenamiento y membrana y también proteínas como fases sortivas separadas (Armitage et al. 2013). pp-Lfer es un enfoque muy adecuado para modelar estos procesos de sorción o partición y los resultados para dos grandes conjuntos de datos se presentan en la Tabla 3. Los coeficientes e, s, b y v son bastante similares. El único parámetro que es diferente en estos dos modelos es el coeficiente a, que representa la contribución del enlace de hidrógeno (HB) donando propiedades (A) de los químicos en el conjunto de datos. Este efecto tiene sentido porque el grupo fosfato en la estructura de fosfolípidos tiene fuertes propiedades aceptoras de HB. Este ejemplo muestra la fuerza del enfoque PP-Lfer porque representa de cerca el mecanismo de interacciones.
Cuadro 3. LFIs para los coeficientes de partición lípido-agua de almacenamiento (K SL-W) y coeficientes de partición de membrana lípido-agua (K ML-W (liposomas)). Se listan los parámetros (y error estándar), el número de compuestos con los que se calibró el LFER (n), el coeficiente de correlación (r 2) y el error estándar de estimación (SE). log K = c + e E + s S + a A + b B + v V.
|
Para- medidor |
c |
e |
s |
a |
b |
v |
n |
r 2 |
SE |
|
K SL-W |
-0.07 (0.07) |
0.70 (0.06) |
-1.08 (0.08) |
-1.72 (0.13) |
-4.14 (0.09) |
4.11 (0.06) |
247 |
0.997 |
0.29 |
|
De (Geisler et al. 2012) |
|||||||||
|
K ML-W (liposomas) |
0.26 (0.08) |
0.85 (0.05) |
-0.75 (0.08) |
0.29 (0.09) |
-3.84 (0.10) |
3.35 (0.09) |
131 |
0.979 |
0.28 |
|
De (Endo et al. 2011) |
|||||||||
K SL-W: los coeficientes de reparto lipídico de almacenamiento son valores medios para diferentes tipos de aceite. Datos brutos y PP-Lfer (para 37 o C) reportados en (Geisler et al. 2012).
K ML-W (liposomas): datos de liposomas compuestos por fosfatidilcolina (PC) o PC mezclados con otros lípidos de membrana. Datos brutos (20-40 o C) y PP-Lfer reportados en (Endo et al. 2011).
Ejemplos de QSPR para sorción al suelo
Numerosos QSPR están disponibles para la sorción del suelo (ver sección sobre Sorción). También el coeficiente de sorción normalizado de carbono orgánico (K oc) se relaciona linealmente con el coeficiente de partición octanol-agua (ver Figura 5).

El modelo de la Figura 5 sólo es válido para químicos orgánicos hidrófobos neutros, no polares, tales como compuestos aromáticos clorados, hidrocarburos aromáticos policíclicos (HAP), bifenilo policlorado (PCB) e insecticidas clorados o, en general, compuestos que solo contienen átomos de carbono, hidrógeno y halógeno. No se aplica a compuestos orgánicos polares e ionizados ni a metales. Para los químicos polares, también otras interacciones pueden influir en la sorción y un enfoque de PP-Lfer también sería útil.
La sorción de los químicos iónicos es más compleja. Para la sorción de compuestos orgánicos catiónicos, los minerales arcillosos pueden ser una fase de sorción igualmente importante como materia orgánica debido a su carga superficial negativa y gran área superficial. La sorción de cationes orgánicos es principalmente un proceso de adsorción que alcanza un máximo a la capacidad de intercambio catiónico (CEC) de una partícula (ver sección Suelo). También los modelos para la predicción de sorción de compuestos catiónicos son más complicados y recientemente se han realizado primeros intentos (Droge y Goss, 2013). El principal mecanismo de sorción para productos químicos aniónicos es la sorción en materia orgánica. El coeficiente de sorción de los químicos aniónicos es sustancialmente menor que para la forma neutra del químico, aproximadamente un factor 10-100 para K OC (Tülp et al. 2009). En caso de productos químicos débilmente disociantes como los ácidos carboxílicos, el coeficiente de sorción a menudo se puede estimar a partir del coeficiente de sorción de la forma no iónica y la fracción del químico que está presente en la forma no ionizada (ver sección Propiedades químicas relevantes).
Fiabilidad y limitaciones de QSPR
Los modelos predictivos tienen limitaciones y es importante conocerlas. No hay un solo modelo que pueda predecir un parámetro para todos los químicos. Cada modelo tendrá un dominio de aplicabilidad y es importante aplicar un modelo solo a una sustancia química dentro de ese dominio. Por lo tanto, se tiene que definir una orientación sobre cómo seleccionar un modelo específico. También es importante darse cuenta de que en muchos programas de computadora (como los programas de modelado del destino), las estimaciones y predicciones se incorporan implícitamente en estos programas.
Otro aspecto es la fiabilidad de la predicción. El modelo en sí puede mostrar un buen ajuste (alto r 2) para el conjunto de entrenamiento (los químicos utilizados para desarrollar el modelo), pero la confiabilidad real debe probarse con un conjunto separado de productos químicos (el conjunto de validación) y se pueden aplicar una serie de procedimientos estadísticos para probar la precisión y predictivo potencia del modelo. La OCDE ha desarrollado un conjunto de reglas que deben aplicarse en la validación de los modelos QSPR y QSAR.
Referencias
Abraham, M.H. (1993). Escalas de enlace de hidrógeno de soluto - su construcción y aplicación a procesos fisicoquímicos y bioquímicos. Chemical Society Opiniones 22, 73-83.
Armitage, J.M., Arnot, J.A., Wania, F., Mackay, D. (2013). Desarrollo y evaluación de un modelo mecanicista de bioconcentración para químicos orgánicos ionogénicos en peces. Toxicología y Química Ambiental 32, 115-128.
Bruggeman, W.A., Opperhuizen, A., Wijbenga, A., Hutzinger, O. (1984). Bioacumulación de sustancias químicas súper lipofílicas en peces. Química Toxicológica y Ambiental 7, 173-189.
Droge, S.T.J., Goss, K.U. (2013). Desarrollo y evaluación de un nuevo modelo de sorción para cationes orgánicos en suelo: Aportes de materia orgánica y minerales arcillosos. Ciencia y Tecnología Ambiental 47, 14233-14241.
Endo, S., Escher, B.I., Goss, K.U. (2011). Capacidades de los lípidos de membrana para acumular químicos orgánicos neutros. Ciencia y Tecnología Ambiental 45, 5912-5921.
Endo, S., Goss, K.U. (2014). Aplicaciones de las relaciones energéticas libres lineales poliparamétricas en la química ambiental. Ciencia y Tecnología Ambiental 48, 12477-12491.
Eriksson, L., Hermens, J.L.M., Johansson, E., Verhaar, H.J.M., Wold, S. (1995). Análisis multivariado de datos de toxicidad acuática con pls. Ciencias Acuáticas 57:217-241.
Geisler, A., Endo, S., Goss, K.U. (2012). Repartición de químicos orgánicos a lípidos de almacenamiento: dilucidar la dependencia de la composición de ácidos grasos y la temperatura. Ciencia y Tecnología Ambiental 46, 9519-9524.
Goss, K.-U., Schwarzenbach, R.P. (2001). Relaciones lineales de energía libre utilizadas para evaluar el reparto en equilibrio de compuestos orgánicos. Ciencia y Tecnología Ambiental 35, 1-9.
Goss, K.U., Schwarzenbach, R.P. (2003). Reglas generales para evaluar el reparto en equilibrio de compuestos orgánicos: Éxitos y trampas. Revista de Educación Química 80, 450-455.
Hansch, C., Streich, M., Geiger, F., Muir, R.M., Maloney, P.P., Fujita, T. (1963). Correlación de la actividad biológica de reguladores del crecimiento vegetal y derivados de cloromicetina con constantes de hammett y coeficientes de partición. Revista de la Sociedad Americana de Química 85, 2817-&.
Jonker, M.T.O., van der Heijden, S.A. (2007). Corte de hidrofobicidad del factor de bioconcentración: Un fenómeno artificial reconstruido. Ciencia y Tecnología Ambiental 41, 7363-7369.
Katritzky, A.R., Slavov, S., Radzvilovits, M., Stoyanova-Slavova, I., Karelson, M. (2009). Enfoques de química computacional para entender cómo la estructura determina las propiedades. Zeitschrift Fur Naturforschung Sección B-a Revista de Ciencias Químicas 64:773-777.
Könemann, H., Van Leeuwen, K. (1980). Toxicocinética en peces: Acumulación y eliminación de seis clorobencenos por guppies. Quimosfera 9, 3-19.
Kraaij, R., Mayer, P., Busser, F.J.M., Bolscher, M.V., Seinen, W., Peajes, J. (2003). Las concentraciones medidas de poro-agua hacen que el reparto de equilibrio funcione, un análisis de datos. Ciencia y Tecnología Ambiental 37, 268-274.
Sabljic, A., Güsten, H., Verhaar, H.J.M., Hermens, J.L.M. (1995). Modelado Qsar de la sorción de suelos. Mejoras y sistemáticas de las correlaciones log k oc vs log k ow. Quimosfera 31, 4489-4514.
Tülp, H.C., Fenner, K., Schwarzenbach, R.P., Goss, K.U. (2009). sorción dependiente del pH de químicos orgánicos ácidos a materia orgánica del suelo. Ciencia y Tecnología Ambiental 43, 9189-9195.
Veith, G.D., Defoe, D.L., Bergstedt, B.V. (1979). Medición y estimación del factor de bioconcentración de químicos en peces. Revista de la Junta de Investigación Pesquera de Canadá 36, 1040-1048.
¿Qué es un QSPR y por qué es útil?
Qué técnicas se aplican para derivar un QSPR.
¿Qué parámetros químicos se aplican en un QSPR?