
4.2: Toxicodinámica e interacciones moleculares
- Page ID
- 70490

4.2. Toxicodinámica e interacciones moleculares
Autor: Timo Hamers
Críticos: Frank van Belleghem y Ludek Blaha
Objetivos de aprendizaje
Deberías ser capaz de
- explicar que una respuesta tóxica requiere una interacción molecular entre un compuesto tóxico y su diana
- nombrar al menos tres tipos diferentes de dianas biomoleculares
- nombrar al menos tres funciones de proteínas que pueden verse obstaculizadas por compuestos tóxicos
- explicar en términos generales las consecuencias de la interacción molecular con una proteína receptora, una enzima, una proteína transportadora, una molécula de ADN y una bicapa lipídica de membrana.
Palabras clave: Receptor; Factor de transcripción; Aductos de ADN; Membrana; Estrés oxidativo
Descripción
La toxicodinámica describe las interacciones dinámicas entre un compuesto y su diana biológica, conduciendo finalmente a un efecto (adverso). En este Capítulo 4.2, se ha descrito la toxicodinámica para procesos que conducen a diversos efectos adversos. Cualquier efecto adverso por una sustancia tóxica es el resultado de una interacción entre el tóxico y su diana biomolecular (es decir, mecanismo de acción). Las dianas biomoleculares incluyen una proteína, una molécula de ADN o ARN, una membrana bicapa de fosfolípidos, pero también moléculas pequeñas que tienen funciones específicas para mantener la homeostasis celular.
Tanto los compuestos endógenos como los xenobióticos que se unen a proteínas se denominan ligandos. La consecuencia de una interacción proteica depende del papel de la proteína diana, p.
1. Receptor
2. Enzima
3. Proteína
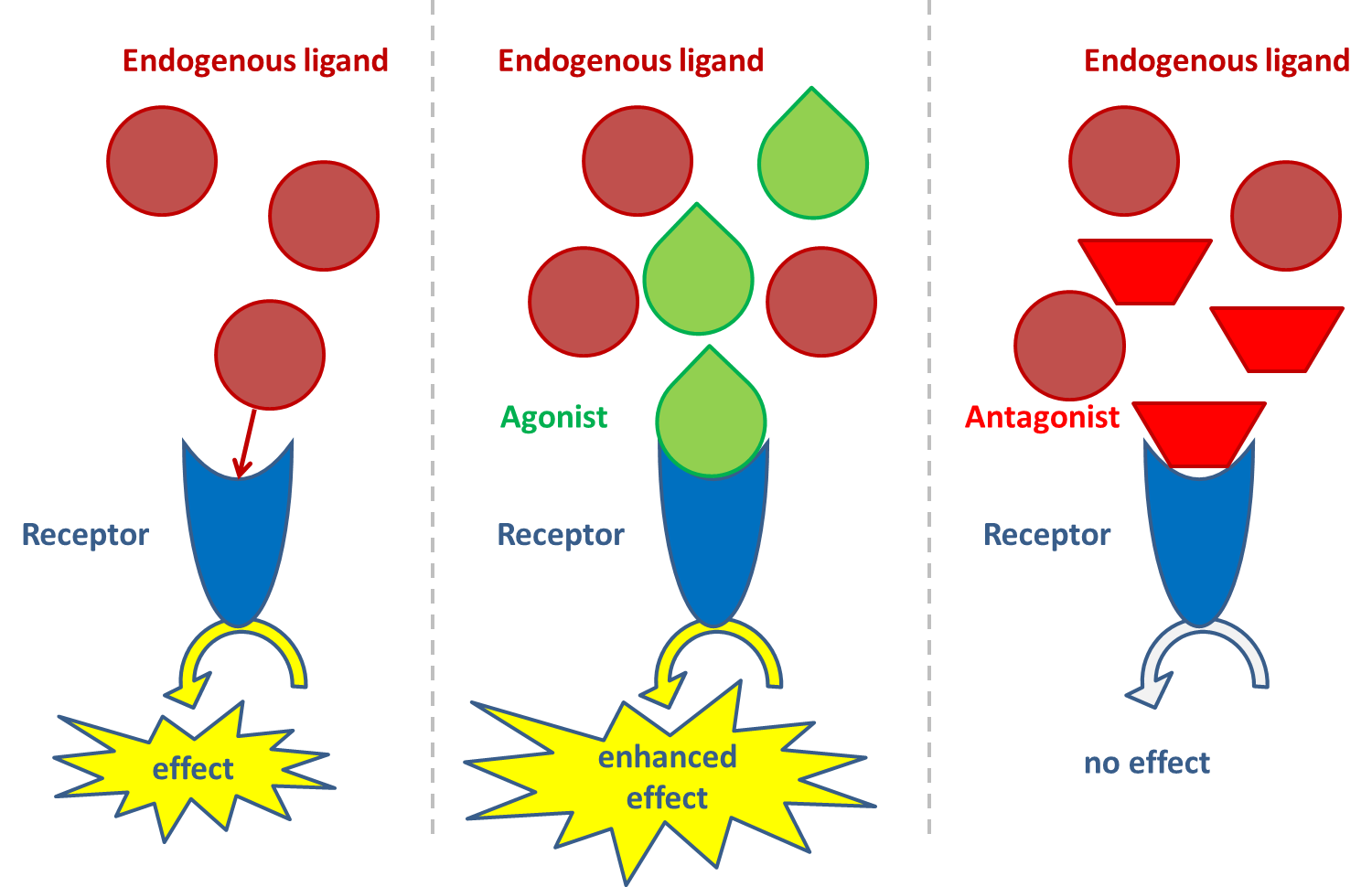
Las proteínas receptoras se unen específicamente y responden a ligandos de señalización endógenos tales como hormonas, prostaglandinas, factores de crecimiento o neurotransmisores, causando una respuesta celular típica. Las proteínas receptoras pueden localizarse en la membrana celular, en el citosol y en el núcleo de una célula. Los ligandos agonistas del receptor activan la proteína receptora mientras que los ligandos antagonistas inactivan el receptor y evitan que los agonistas (endógenos) activen el receptor. Basándose en el papel de la proteína receptora, la unión por ligandos puede interferir con canales iónicos, receptores acoplados a proteínas G, receptores unidos a enzimas o receptores nucleares. Los ligandos xenobióticos pueden interferir con estas respuestas celulares actuando como ligandos agonistas o antagonistas (enlace a la sección sobre la interacción del Receptor).
Los compuestos que se unen a una enzima generalmente causan inhibición de la actividad enzimática, es decir, una disminución en la tasa de conversión del sustrato (s) endógeno (s) de la enzima en su/su (s) producto (s) correspondiente (s). Los compuestos que se unen no covalentemente a una enzima causan inhibición reversible, mientras que los compuestos que se unen covalentemente a una enzima causan inhibición irreversible (enlace a la sección sobre inactivación de proteínas).
De manera similar, los compuestos que se unen a una proteína transportadora suelen inhibir el transporte del ligando endógeno natural. Dichas proteínas transportadoras pueden ser responsables del transporte local de ligandos endógenos a través de la membrana celular, pero también del transporte periférico de ligandos endógenos a través de la sangre de un órgano al otro (enlace a la sección Interrupción endocrina).
Además de la interacción con proteínas receptoras, enzimáticas o transportadoras funcionales, los compuestos tóxicos también pueden interactuar con proteínas estructurales. Por ejemplo, el citoesqueleto puede ser dañado por compuestos tóxicos que bloquean la polimerización de actina, evitando así la formación de filamentos.
Además de las proteínas, las macromoléculas de ADN y ARN pueden ser dianas para la unión de compuestos. Especialmente la base guanina puede unirse covalentemente por compuestos electrofílicos, tales como metabolitos reactivos. Dichos aductos de ADN pueden causar errores de copia durante la replicación del ADN que conducen a mutaciones puntuales (enlace a la sección sobre Genotoxicidad).
Los compuestos también pueden interferir con las membranas bicapa fosfolipídicas, especialmente con la membrana celular externa y con las membranas mitocondriales. Los compuestos perturban la integridad y funcionamiento de la membrana dividiendo en la bicapa lipídica. La pérdida de integridad de la membrana puede conducir en última instancia a fugas de electrolitos y pérdida de potencial de membrana.
|
La partición en la bicapa lipídica es un proceso inespecífico. Por lo tanto, las concentraciones en membranas biológicas que provocan efectos a través de este modo de acción no difieren entre los compuestos. Como tal, este tipo de toxicidad es considerada como una “toxicidad basal” (también llamada “narcosis”), la cual es ejercida por todos los químicos. Por ejemplo, la concentración química en una membrana diana que causa 50% de mortalidad en una población de prueba es de alrededor de 50 mmol/kg de lípido, independientemente de la especie o compuesto en consideración. Sin embargo, con base en los niveles de exposición externa, los compuestos tienen diferentes potencias narcóticas. Después de todo, para alcanzar concentraciones internas similares basadas en lípidos, se requieren diferentes concentraciones de exposición, dependiendo del coeficiente de reparto lípido-agua, que es una propiedad intrínseca de un compuesto, y no de la especie. La acción narcótica no es el único mecanismo por el cual los compuestos pueden dañar la integridad de la membrana Los compuestos llamados “ionóforos”, por ejemplo, actúan como portadores de iones que transportan iones a través de la membrana, interrumpiendo así el gradiente de electrolitos a través de la membrana. Los ionóforos no deben confundirse con compuestos que abren o cierran los canales iónicos, aunque ambos tipos de compuestos pueden alterar el gradiente electrolítico a través de la membrana. La diferencia es que los ionóforos se disuelven en la membrana bicapa y transportan iones transportando a través de la propia membrana, mientras que los inhibidores o estimuladores de canales iónicos cierran o abren, respectivamente, un canal proteico en la membrana que actúa como puerta para el transporte iónico. |
Finalmente, cabe mencionar aquí que algunos compuestos pueden causar estrés oxidativo al aumentar la formación de especies reactivas de oxígeno (ROS), como H 2 O 2, O 3, O 2 •-, •OH, NO• o RO•. Las ROS son metabolitos de oxígeno que se encuentran en cualquier organismo vivo aeróbico. Los compuestos pueden causar directamente un aumento en la formación de ROS al someterse a ciclos redox o interferir con la cadena de transporte de electrones. Alternativamente, los compuestos pueden causar un aumento indirecto en la formación de ROS por interferencia con antioxidantes eliminadores de ROS, que van desde moléculas pequeñas (por ejemplo, glutatión) hasta proteínas (por ejemplo, catalasa o superóxido dismutasa). Para los compuestos que causan estrés oxidativo directo o indirecto, no es el compuesto en sí el que tiene una interacción molecular con la diana, sino las ROS las que pueden unirse covalentemente al ADN, las proteínas y los lípidos (enlace a la sección sobre Estrés Oxidativo).
Nombrar tres dianas biomoleculares que puedan verse afectadas por un compuesto
Nombra tres mecanismos diferentes por los cuales un compuesto puede afectar el transporte de analitos a través de la membrana celular
¿Cuál es la diferencia entre un agonista del receptor y un antagonista del receptor?
4.2.1. Inactivación de proteínas
Autor: Timo Hamers
Críticos: Frank van Belleghem y Ludek Blaha
Objetivos de aprendizaje:
Deberías ser capaz de
- discutir cómo un compuesto que se une a una proteína puede inhibir la unión del ligando y, por lo tanto, obstaculizar la función de la proteína
- explicar el mecanismo de acción de los insecticidas organofosforados inhibidores de la acetilcolinesterasa
- explicar el mecanismo de acción de los fenoles halogenados que inhiben el transporte de la hormona tiroidea por la transtiretina
- distinguir entre inactivación de proteínas reversible e irreversible
- distinguir entre inhibición enzimática competitiva, no competitiva y no competitiva
Palabras clave: inhibición enzimática; acetilcolinesterasa, transtiretina, inhibición competitiva, inhibición no competitiva, inhibición no competitiva
Introducción
Las proteínas juegan un papel importante en los procesos bioquímicos esenciales, incluida la catálisis de reacciones metabólicas, la replicación y reparación del ADN, el transporte de mensajeros (por ejemplo, hormonas) o las respuestas de los receptores a dichos mensajeros. Muchos compuestos tóxicos ejercen su acción tóxica al unirse a una proteína y, por lo tanto, perturbar estas funciones vitales de la proteína.
Inhibición de la función de transporte de proteínas
La unión de compuestos xenobióticos a una proteína transportadora puede dificultar la unión del ligando natural de la proteína, inhibiendo así la función transportadora de la proteína. Un ejemplo de tal inhibición es la unión de fenoles halogenados a transtiretina (TTR). La TTR es una proteína de transporte de hormonas tiroideas, presente en la sangre. Tiene dos lugares de unión para el transporte de la hormona tiroidea, es decir, principalmente tiroxina (T4) en mamíferos y principalmente triyodotironina (T3) en otros vertebrados (Figura 1). Los compuestos con alto parecido estructural con la hormona tiroidea (especialmente los fenoles halogenados, como los metabolitos hidroxilados de PCB o PBDE), son capaces de competir con la hormona tiroidea por la unión a TTR. Aparte de que esto mejora la distribución de los compuestos tóxicos, esto también provoca un aumento de la hormona tiroidea no unida en la sangre, que luego está disponible gratuitamente para su absorción en el hígado, conjugación metabólica y excreción urinaria. En última instancia, esto puede llevar a disminuir los niveles de hormona tiroidea en la sangre.
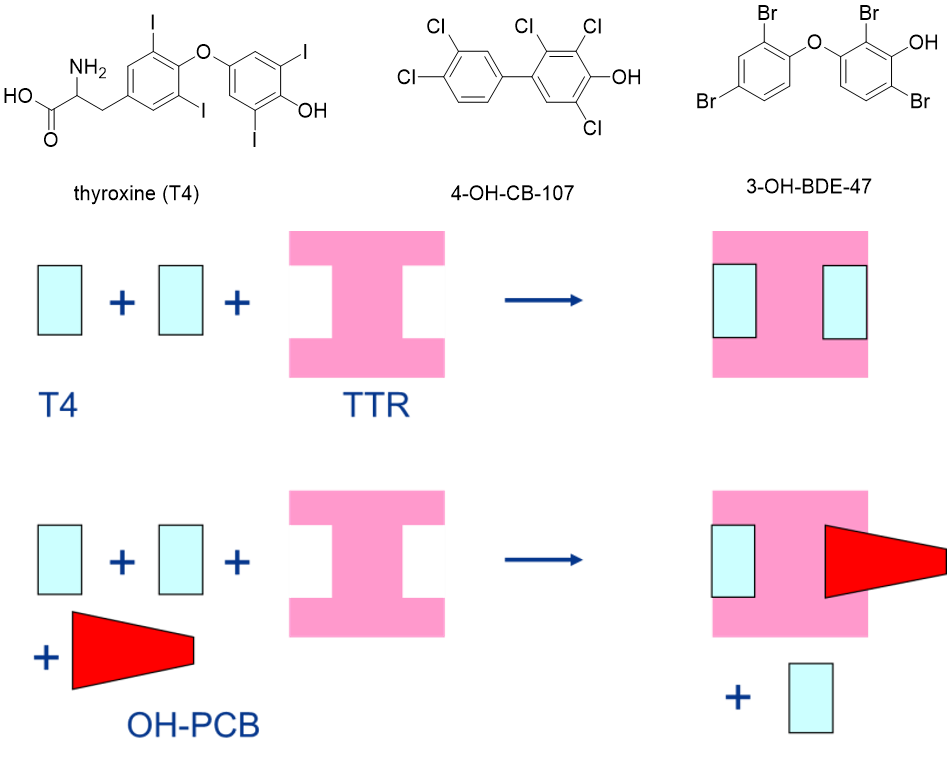
Inhibición de la actividad enzimática de la proteína
Las proteínas involucradas en la catálisis de una reacción metabólica se llaman enzimas. La fórmula general de tal reacción es
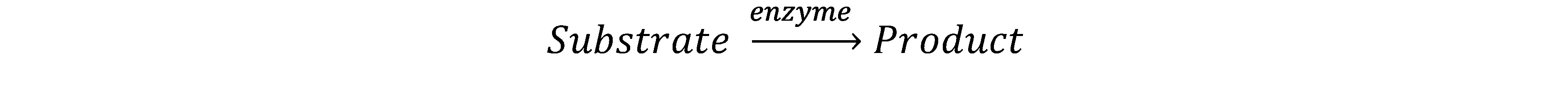
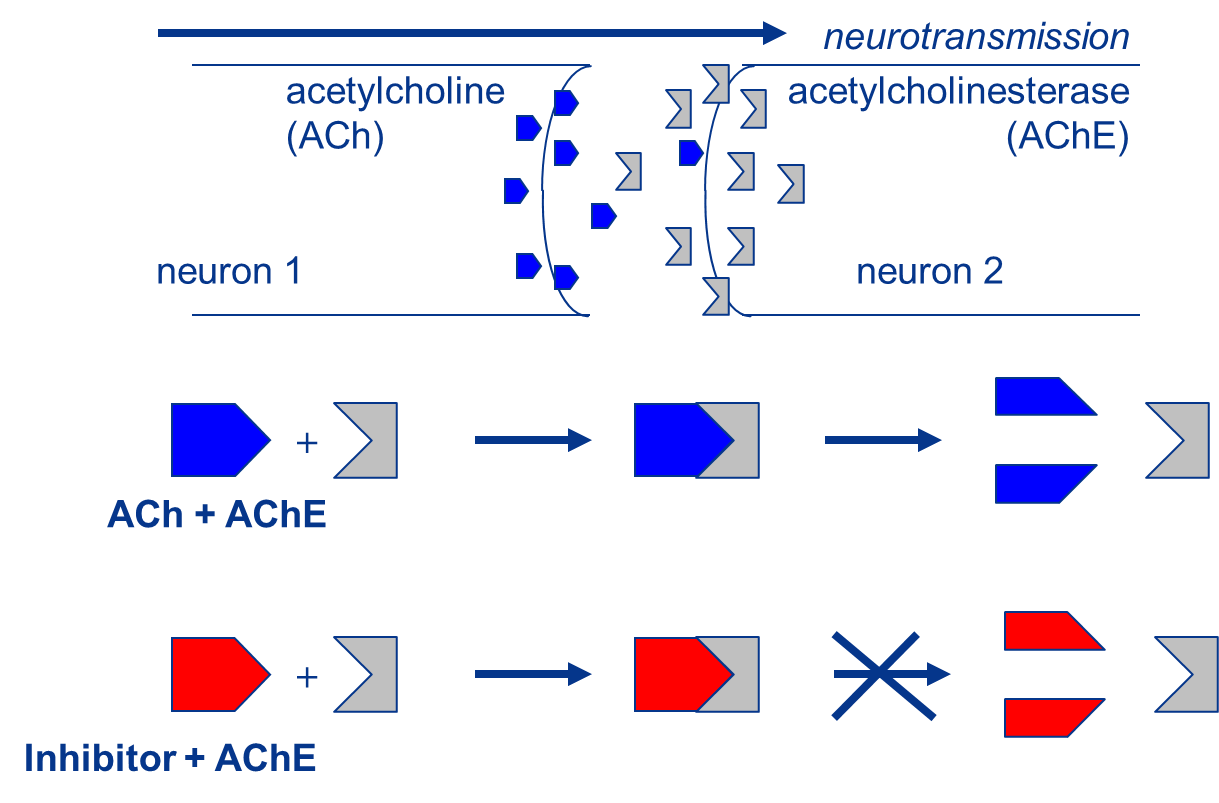
La unión de un compuesto tóxico a una enzima generalmente causa una inhibición de la actividad enzimática, es decir, una disminución en la tasa de conversión del sustrato o sustratos endógenos de la enzima en su/sus productos correspondientes. En la práctica, esto provoca una respuesta tóxica por un excedente de sustrato y/o un déficit de producto. Uno de los ejemplos clásicos de inhibición enzimática por compuestos tóxicos es la inhibición de la enzima acetilcolinesterasa (AChE) por insecticidas organofosforados. La AChE cataliza la hidrólisis del neurotransmisor acetilcolina (ACh), en las sinapsis colinérgicas. Durante la transferencia de un potencial de acción de una célula a otra, se libera ACh en estas sinapsis de la célula presináptica a la hendidura sináptica con el fin de estimular el receptor de acetilcolina (AChR) en la membrana de la célula postsináptica. La AChE, que también está presente en estas sinapsis, es entonces responsable de descomponer la ACh en ácido acético y colina:

Mediante la unión covalente a residuos de serina en el sitio activo de la enzima AChE, los insecticidas organofosforados pueden inhibir esta reacción provocando la acumulación del neurotransmisor ACh en la sinapsis (Fig. 2). Como consecuencia, el AChR es sobreestimulado causando convulsiones, hipertensión, debilidad muscular, salivación, lagrimeo, problemas gastrointestinales y latidos cardíacos lentos.
Inhibición enzimática irreversible vs reversible
Los insecticidas organofosforados se unen covalentemente a la enzima AChE provocando una inhibición irreversible de la enzima. La inhibición enzimática irreversible aumenta progresivamente en el tiempo después de la cinética de primer orden (enlace a la sección sobre Bioacumulación y modelado cinético). La recuperación de la actividad enzimática solo se puede obtener mediante la síntesis de novo de enzimas. A diferencia de la inhibición de AChE, la inhibición de la función de transporte de T4 de TTR es reversible porque los fenoles halogenados se unen a TTR de manera no covalente. De manera similar, la unión no covalente de un compuesto tóxico a una enzima provoca una inhibición reversible de la actividad enzimática.
Además de la unión enzimática covalente y no covalente, la inhibición enzimática irreversible puede ocurrir cuando los compuestos tóxicos causan un error durante la síntesis enzimática. Por ejemplo, los iones de metales esenciales, que están presentes como cofactores en el sitio activo de muchas enzimas, pueden ser reemplazados por iones de otros metales durante la síntesis enzimática, produciendo enzimas inactivas. Un ejemplo clásico de tal disminución de la actividad enzimática es la inhibición de ácido δ-aminolevulínico deshidratasa (δ-ALAD) por plomo. En este caso, el plomo reemplaza al zinc en el sitio activo de la enzima, inhibiendo así una etapa catalítica en la síntesis de un precursor del hemo, un cofactor de la proteína hemoglobulina (enlace a la sección sobre Mecanismos de Toxicidad de los metales).
Con respecto a la inhibición enzimática reversible, se pueden distinguir tres tipos de inhibición, es decir, inhibición competitiva, no competitiva y no competitiva (Figura 3).
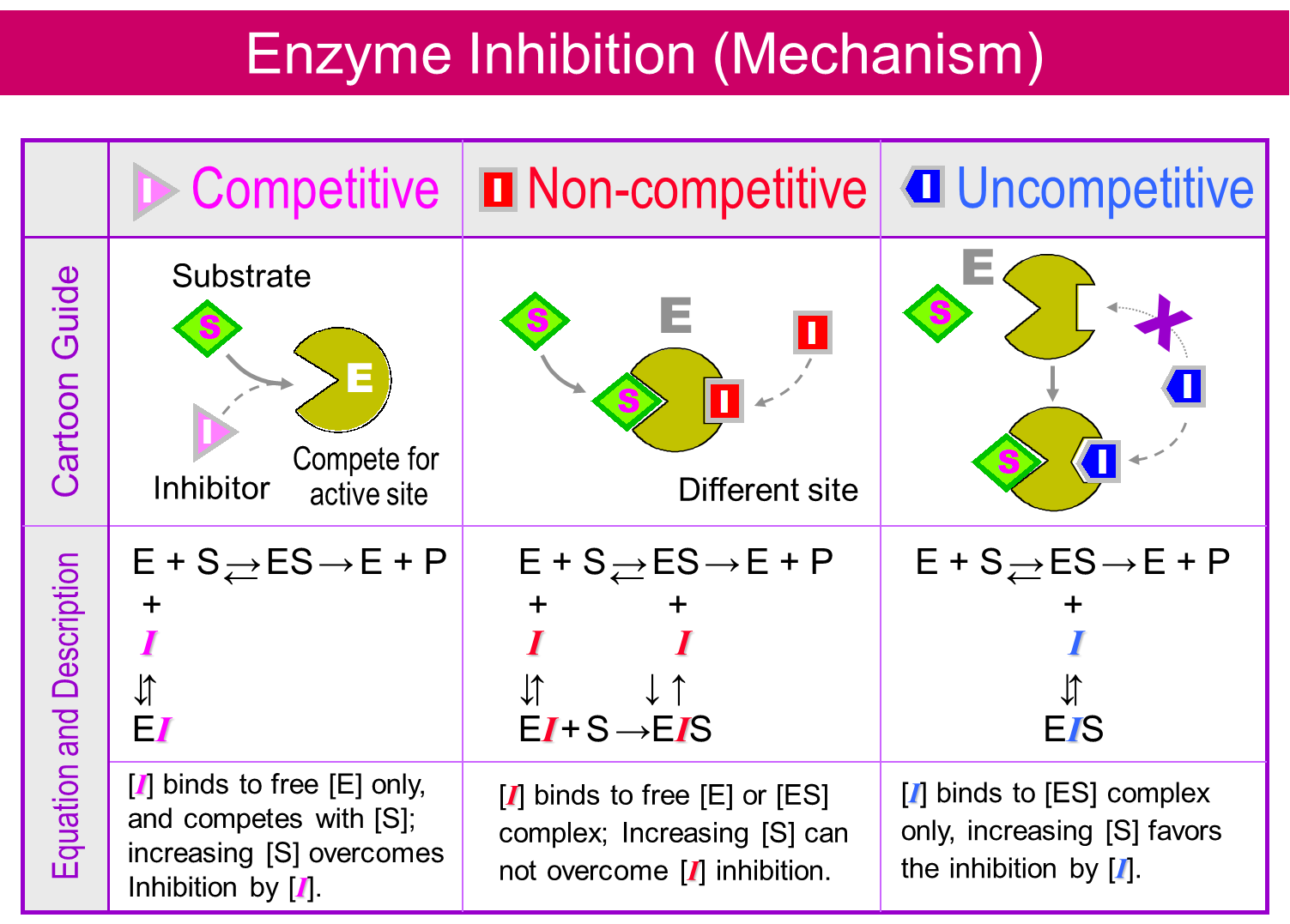
La inhibición competitiva se refiere a una situación en la que el químico compite (“lucha”) con el sustrato por unirse al sitio activo de la enzima. La inhibición competitiva es muy específica, ya que requiere que el inhibidor se asemeje al sustrato y encaje en el mismo bolsillo de unión del sitio activo. El ejemplo de unión a TTR descrito anteriormente es un ejemplo típico de inhibición competitiva entre la hormona tiroidea y los fenoles halogenados para la ocupación del sitio de unión a TTR. Un ejemplo más clásico de inhibición competitiva es la inhibición de la beta-lactamasa por la penicilina. La beta-lactamasa es una enzima responsable de la hidrólisis de la beta-lactama, que es el paso final en la síntesis de la pared celular bacteriana. Por síntesis defectuosa de la pared celular, la penicilina es un antibiótico que causa la muerte bacteriana.
La inhibición no competitiva se refiere a una situación en la que el químico se une a un sitio alostérico de la enzima (es decir, no al sitio activo), lo que provoca un cambio conformacional del sitio activo. Como consecuencia, el sustrato no puede ingresar al sitio activo, o el sitio activo se vuelve inactivo, o el producto no puede liberarse del sitio activo. Por ejemplo, los fármacos antifúngicos de equinocandina inhiben de manera no competitiva la enzima 1,3-beta glucano sintasa, que es responsable de la síntesis de beta-glucano, un constituyente principal de la pared celular fúngica. La falta de beta-glucano en las paredes celulares fúngicas impide la resistencia fúngica contra las fuerzas osmóticas, lo que lleva a la lisis celular.
La inhibición no competitiva se refiere a una situación en la que el químico solo puede unirse a la enzima si el sustrato se une simultáneamente. La unión al sustrato conduce a un cambio conformacional de la enzima, lo que conduce a la formación de un sitio de unión alostérico para el inhibidor. La inhibición no competitiva es más común en reacciones enzimáticas de dos sustratos que en reacciones enzimáticas de un sustrato. Un ejemplo de inhibición no competitiva es la inhibición por litio de la enzima inositol mono fosfatasa (IMPasa), la cual está involucrada en el reciclaje del segundo mensajero inositol-3-fosfato (I3P) (enlace a la sección sobre la interacción del Receptor). IMPase está involucrada en la etapa final de desfosforilar inositol monofosfato en inositol. Dado que el litio es el tratamiento primario para el trastorno bipolar, esta observación ha llevado a la hipótesis de agotamiento del inositol de que la inhibición del metabolismo del inositol fosfato ofrece una explicación plausible de los efectos terapéuticos del litio.
Explicar cómo la unión de los insecticidas organofosforados a las enzimas acetilcolinesterasas puede causar neurotoxicidad.
Explicar cómo los fenoles organohalogenados pueden causar disminución de los niveles sanguíneos de la hormona tiroidea T4.
¿Cuál es la diferencia entre un inhibidor enzimático competitivo y uno no competitivo?
¿Es posible superar a un inhibidor enzimático competitivo aumentando la concentración de sustrato?
¿Es posible superar a un inhibidor enzimático no competitivo aumentando la concentración de sustrato?
4.2.2. Interacción del receptor
Autor: Timo Hamers
Críticos: Frank van Belleghem y Ludek Blaha
Objetivos de aprendizaje
Deberías ser capaz de
- explicar los posibles efectos de la interferencia compuesta con los canales iónicos.
- explicar los posibles efectos de la interferencia de compuestos con receptores acoplados a proteínas G (GPCR).
- explicar los posibles efectos de la interferencia de compuestos con receptores ligados a enzimas.
- explicar los posibles efectos de la interferencia compuesta con los receptores nucleares.
- comprender qué son las vías de señalización y cómo pueden verse afectadas por compuestos tóxicos
Palabras clave: Canales iónicos, receptores acoplados a proteínas G, receptores enlazados a enzimas, receptores nucleares
Introducción
Las proteínas receptoras se unen específicamente y responden a ligandos de señalización endógenos tales como hormonas, prostaglandinas, factores de crecimiento o neurotransmisores, causando una respuesta celular típica. Las proteínas receptoras pueden localizarse en la membrana celular, en el citosol y en el núcleo de una célula. Los ligandos agonistas del receptor activan la proteína receptora mientras que los ligandos antagonistas inactivan el receptor y evitan que los agonistas (endógenos) activen el receptor (Figura 1). Basándose en el papel de la proteína receptora, la unión por ligandos puede interferir con:
1. canales iónicos
2. Receptores acoplados a proteína G
3. Receptores ligados a enzimas
4. receptores nucleares.
Los ligandos xenobióticos pueden interferir con estas respuestas celulares actuando como ligandos agonísticos o antagonistas.
.
1. Canales de iones
Los canales iónicos son complejos proteicos transmembrana que transportan iones a través de una membrana bicapa de fosfolípidos. Los canales iónicos son especialmente importantes en la neurotransmisión, cuando los neurotransmisores estimulantes (por ejemplo, acetilcolina o ACh) se unen a la parte del receptor (llamado ionotrópico) del canal iónico y abren el canal iónico por un período de tiempo muy corto (es decir, milisegundos). Como resultado, los iones pueden atravesar la membrana provocando un cambio en el potencial transmembrana (ver Figura). Por otro lado, la unión al receptor mediante la inhibición de neurotransmisores (por ejemplo, ácido gamma-aminobutírico o GABA) impide la apertura de canales iónicos.
Los compuestos que interfieren con los canales de sodio, por ejemplo, son compuestos neurotóxicos (ver sección sobre Neurotoxicidad). Pueden bloquear los canales iónicos o mantenerlos en un estado prolongado o permanentemente abierto. Muchos compuestos que se sabe que interfieren con los canales iónicos son toxinas naturales. Por ejemplo, la tetrodotoxina (TTX), que es producida por bacterias marinas y altamente acumulada en peces globo, y la saxitoxina, que es producida por dinoflagelados y que se acumula en los mariscos, son capaces de bloquear los canales de sodio regulados por voltaje en las células nerviosas. En contraste, la ciguatoxina, que es otra toxina persistente producida por los dinoflagelados que se acumula en peces depredadores posicionados en lo alto de la cadena alimentaria, provoca la prolongación de la apertura de los canales de sodio regulados por voltaje. Algunos pesticidas como el DDT y los insecticidas piretroides también previenen el cierre de los canales de sodio regulados por voltaje en las células nerviosas. Como consecuencia, no se logra la repolarización total del potencial de membrana. En consecuencia, las células nerviosas no alcanzan el potencial de reposo y cualquier nuevo estímulo que sea demasiado bajo para alcanzar el umbral de despolarización en condiciones normales, provocará ahora un nuevo potencial de acción. En otras palabras, las células nerviosas se vuelven hiperexcitables y sufren una serie de potenciales de acción (disparo repetitivo) provocando temblores e hipertermia.
2. Receptores acoplados a proteína G (GPCR)
Los GPCR son receptores transmembrana que transfieren una señal extracelular a una proteína G activada que está conectada al receptor en el lado intracelular de la membrana. Las proteínas G son proteínas heterotrímeras que constan de tres subunidades alfa, beta y gamma, de las cuales la subunidad alfa, en forma inactivada, contiene una molécula de guanosina difosfato (GDP). Al unirse por ligandos endógenos tales como hormonas, prostaglandinas o neurotransmisores (es decir, la señal o “primer mensajero”) al receptor (llamado metabotrópico), un cambio conformacional en el complejo GPCR conduce a un intercambio del GDP por una molécula de guanosina trifosfato (GTP) en el monómero alfa parte de la proteína G, causando la liberación de la subunidad alfa activada de la parte del dímero beta/gamma. El monómero alfa activado puede interactuar con varias enzimas diana provocando un aumento en los “segundos mensajeros” iniciando vías de transducción de señales (ver punto 3 Receptores ligados a enzimas). El complejo beta-gamma restante también puede moverse a lo largo de la superficie interna de la membrana y afectar la actividad de otras proteínas (Figura 2).
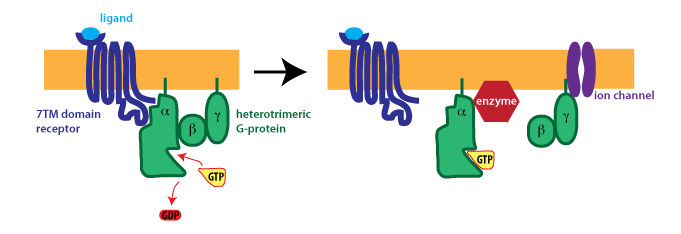
Dos enzimas principales que son activadas por el monómero alfa son la adenilil ciclasa causando un aumento en el AMP cíclico del segundo mensajero (AMPc) y la fosfolipasa C causando un aumento en el segundo mensajero diacilglicerol (DAG). A su vez, el AMPc y DAG activan las proteínas quinasas, que pueden fosforilar muchas otras enzimas. La fosfolipasa C activada también provoca un aumento en los niveles del segundo mensajero inositol-3-fosfato (I3P), que abre canales iónicos en el retículo endoplásmico provocando una liberación de calcio del almacén endoplásmico, que también actúa como segundo mensajero. Por otro lado, el aumento en los niveles de calcio citosólico es templado simultáneamente por el dímero beta/gamma, que puede inhibir los canales de calcio regulados por voltaje en la membrana celular. En última instancia, la señal del GPCR es extinguida por la lenta desfosforilación de GTP en GDP por el monómero alfa activado, provocando que se reorganice con el dímero beta/gamma en la proteína G del trímero inactivado original (ver también cursos.washington.edu/conj/bess/gpcr/gpcr.htm).
El ejemplo más conocido de alteración de la señalización de GPCR es por la toxina del cólera (véase el bloque de texto Toxina del cólera a continuación).
A pesar de la reconocida importancia de los GPRC en medicina y farmacología, hasta ahora se ha prestado poca atención en toxicología a la interacción de xenobióticos con GPCR. Aunque un número limitado de estudios han demostrado que los compuestos alteradores endocrinos, incluidos los HAP, dioxinas, ftalatos, bisfenol-A y DDT pueden interactuar con la señalización de GPCR, las implicaciones toxicológicas de estas interacciones (especialmente con respecto al metabolismo energético alterado) siguen siendo objeto para más investigación (ver revisión de Le Ferrec y Øvrevik, 2018).
|
Toxina del cólera La toxina del cólera es una llamada exotoxina AB de la bacteria Vibrio cholerae, que consiste en una parte A “activa” y una parte B “vinculante” (ver http://www.sumanasinc.com/webcontent/animations/content/diphtheria.html). Al unirse por la parte B a la membrana epitelial intestinal, todo el complejo AB se internaliza en la célula vía endocitosis, y se libera la parte A activa. Esta parte A agrega un grupo ADP-ribosa a las proteínas G haciendo imposible la desfosforilación de GTP de las proteínas G activadas. Como consecuencia, las proteínas G activadas permanecen en un estado activo permanente, la adenilil ciclasa se activa permanentemente y aumentan los niveles de AMPc, lo que a su vez provoca un desequilibrio en la limpieza de iones, es decir, una secreción excesiva de iones cloruro a la luz intestinal y una disminución de la captación de iones sodio desde el lumen intestinal. Debido al aumento de la presión osmótica, se libera agua a la luz intestinal provocando deshidratación y diarrea severa (“heces de agua de arroz”). |
3. Receptores ligados a enzimas
Los receptores ligados a enzimas son receptores transmembrana que transfieren una señal extracelular a una actividad enzimática intracelular. La mayoría de los receptores ligados a enzimas pertenecen a la familia de las proteínas receptoras de tirosina quinasa (RTK). Al unirse por ligandos endógenos tales como hormonas, citocinas o factores de crecimiento (es decir, la señal o mensajero primario) al dominio extracelular de los receptores, los monómeros del receptor se dimerizan y desarrollan actividad quinasa, es decir, se vuelven capaces de acoplarse a un grupo fosfato donado por un donante de alta energía. molécula a una proteína aceptora. El primer sustrato para esta actividad de fosforilación es el propio receptor dimerizado, que acepta un grupo fosfato donado por ATP sobre sus residuos intracelulares de tirosina. Esta autofosforilación es la primera etapa de una vía de señalización que consiste en una cascada de etapas posteriores de fosforilación de otras proteínas cinasas (es decir, transducción de señales), que finalmente conducen a la activación transcripcional de genes seguida de una respuesta celular (Figura 3).
Figura en preparación
Figura 3: Tras la unión del ligando, las proteínas receptoras de tirosina quinasa (TKR) se autofosforilan y pueden fosforilarse (es decir, activar otras proteínas), incluyendo otras quinasas.
Los compuestos xenobióticos pueden interferir con estas vías de señalización de muchas maneras diferentes. Los compuestos pueden evitar la unión del ligando endógeno, bloqueando el receptor o quelando los ligandos endógenos. La mayoría de los inhibidores de RTK inhiben la actividad quinasa directamente actuando como un inhibidor competitivo para la unión de ATP a los residuos de tirosina. Muchos inhibidores de RTK se utilizan en el tratamiento del cáncer, porque la hiperactividad de RTK es típica de muchos tipos de cáncer. Esta sobreactividad puede ser causada, por ejemplo, por niveles aumentados de factores de crecimiento activadores del receptor, o a la dimerización espontánea cuando el receptor está sobreexpresado o mutado).
4. Receptores nucleares
Los receptores nucleares son proteínas que son activadas por compuestos endógenos (a menudo hormonas) que conducen en última instancia a la expresión de genes específicamente regulados por estos receptores. Además de la unión al ligando, la activación de la mayoría de los receptores nucleares requiere dimerización con un factor de transcripción coactivante. Mientras que algunos receptores nucleares se localizan en el núcleo en forma inactiva (por ejemplo, el receptor de la hormona tiroidea), la mayoría de los receptores nucleares se localizan en el citosol, donde se unen a proteínas correpresoras (a menudo proteínas de choque térmico) manteniéndolas en un estado inactivo. Tras la unión del ligando al dominio de unión a ligando (LBD) del receptor, se liberan las proteínas co-represoras y el receptor forma un homodímero con un receptor nuclear activado similar o forma un heterodímero con un receptor nuclear diferente, que a menudo es el receptor retinoide-X (RXR) para nuclear. receptores hormonales. Antes o después de la dimerización, los receptores nucleares activados se translocan al núcleo. En el núcleo, se unen a través de su dominio de unión al ADN (DBD, o “dedo de zinc”) a un elemento sensible en el ADN ubicado en la región promotora de los genes sensibles al receptor. En consecuencia, estos genes se transcriben a ARNm en el núcleo, que se traduce adicionalmente en proteínas en el citoplasma celular, ver Figura 4).
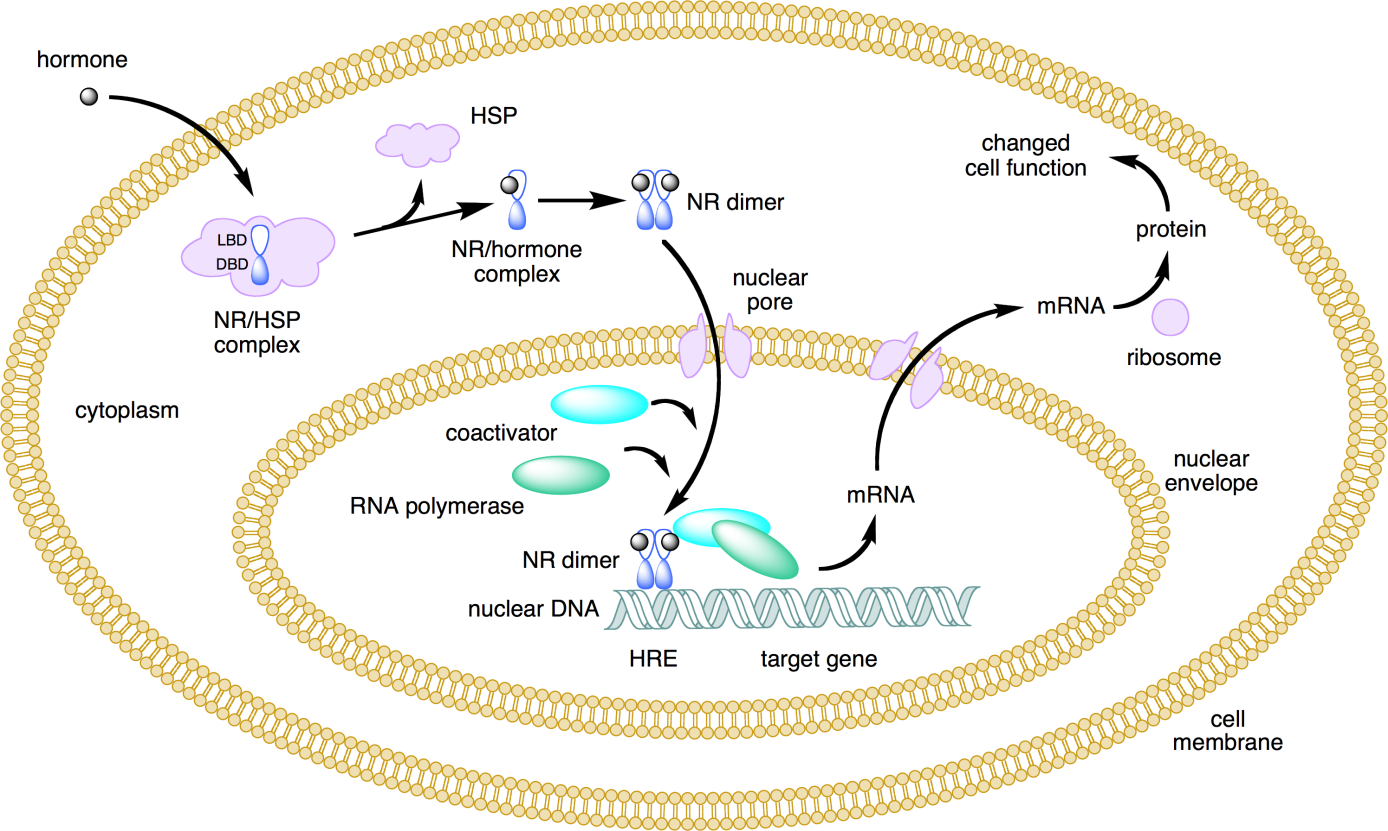
Los compuestos xenobióticos pueden actuar como agonistas o antagonistas de la activación del receptor nuclear. Los químicos que actúan como agonistas del receptor nuclear imitan la acción del (de los) activador (es) endógeno (es), mientras que los químicos que actúan como antagonistas del receptor nuclear básicamente bloquean el LBD del receptor, impidiendo la unión del (de los) activador (es) endógeno (es). En las últimas décadas, la interacción de xenobióticos con receptores nucleares involucrados en la señalización de hormonas esteroides y no esteroideas ha ganado mucha atención de los investigadores que investigan la alteración endocrina (enlace a la sección sobre Interrupción Endocrina). La activación del receptor nuclear también es el mecanismo clave en la toxicidad similar a las dioxinas (véase el bloque de texto toxicidad similar a las dioxinas a continuación).
|
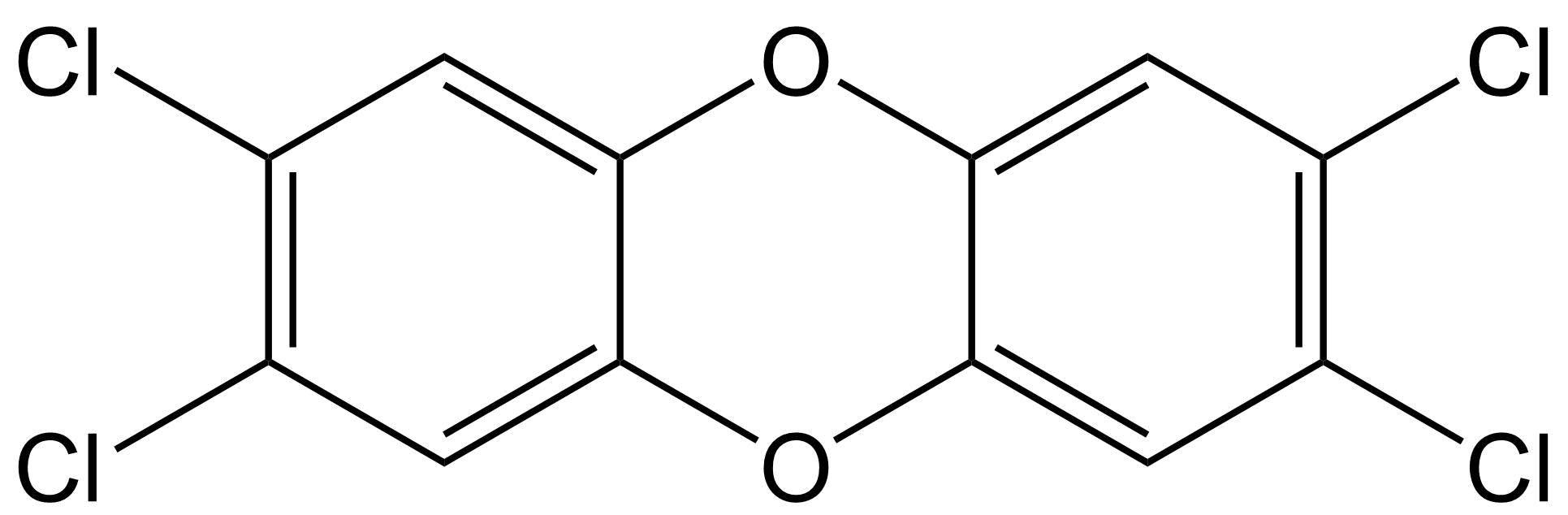
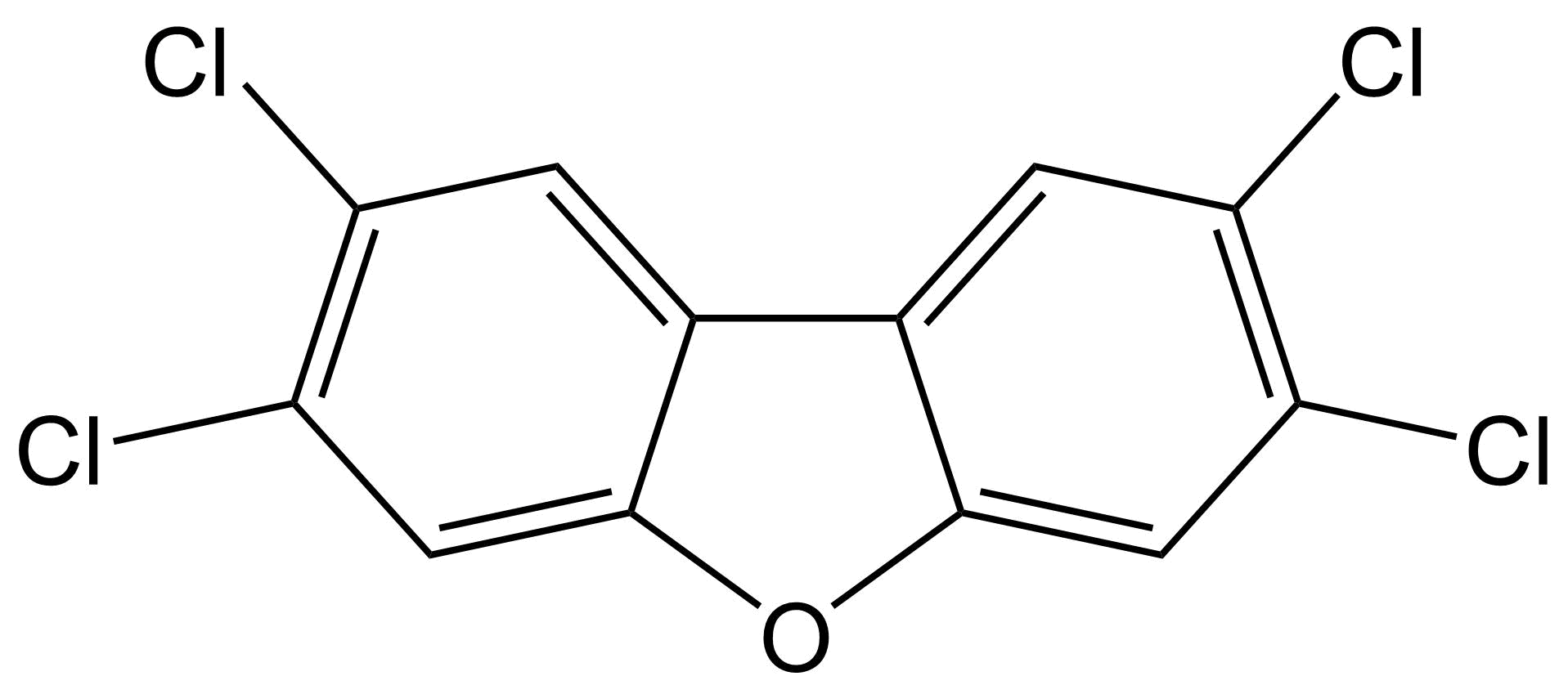
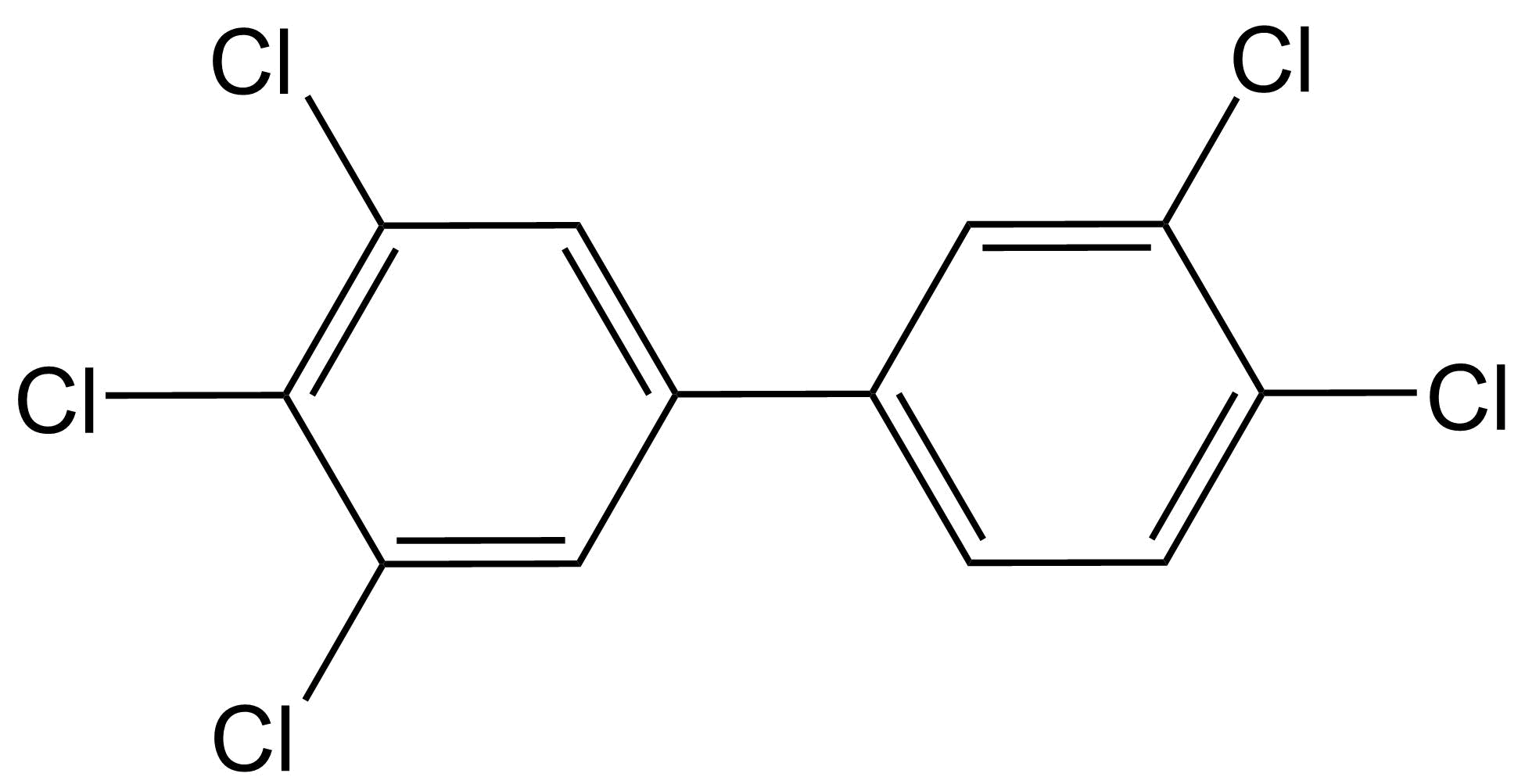
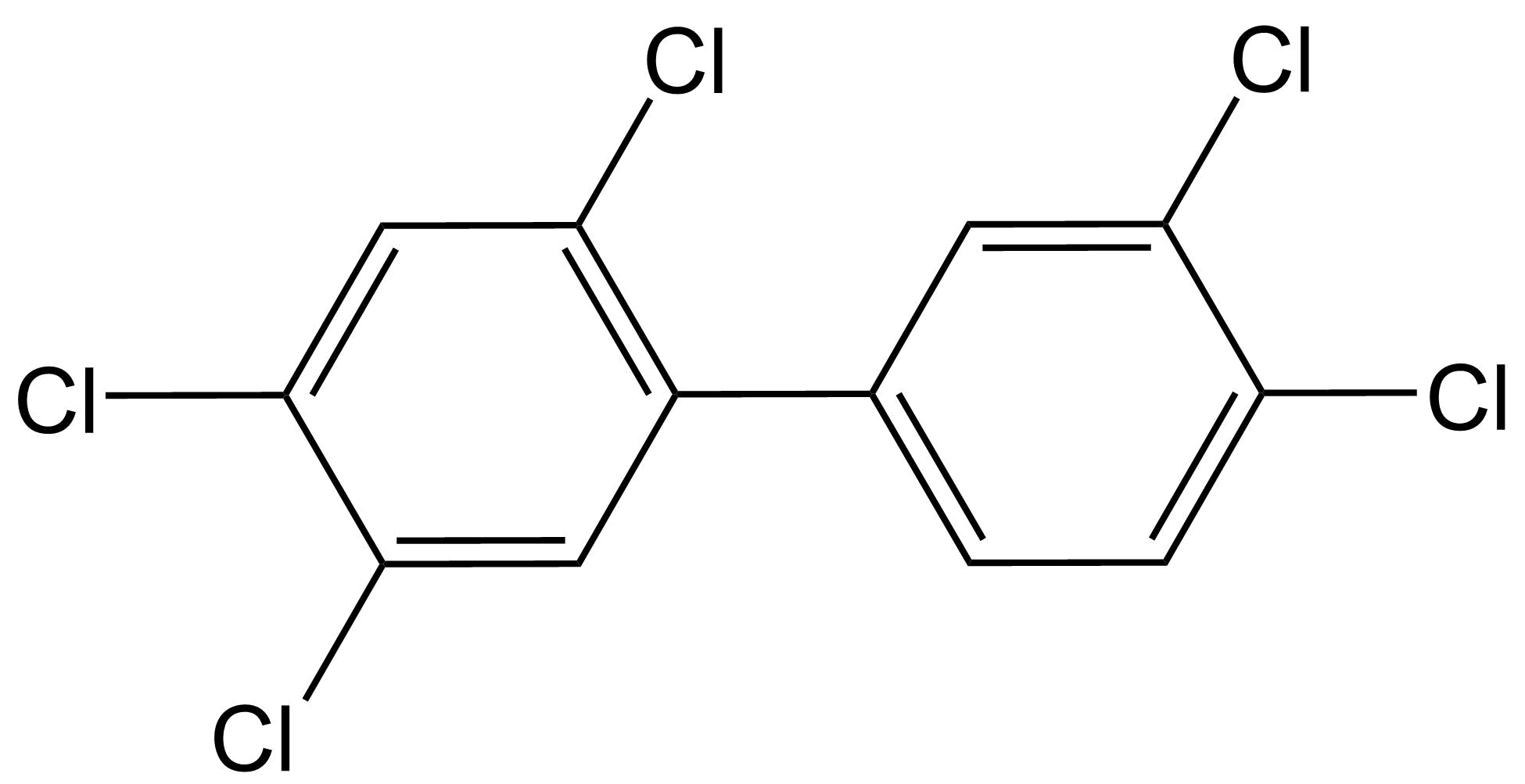
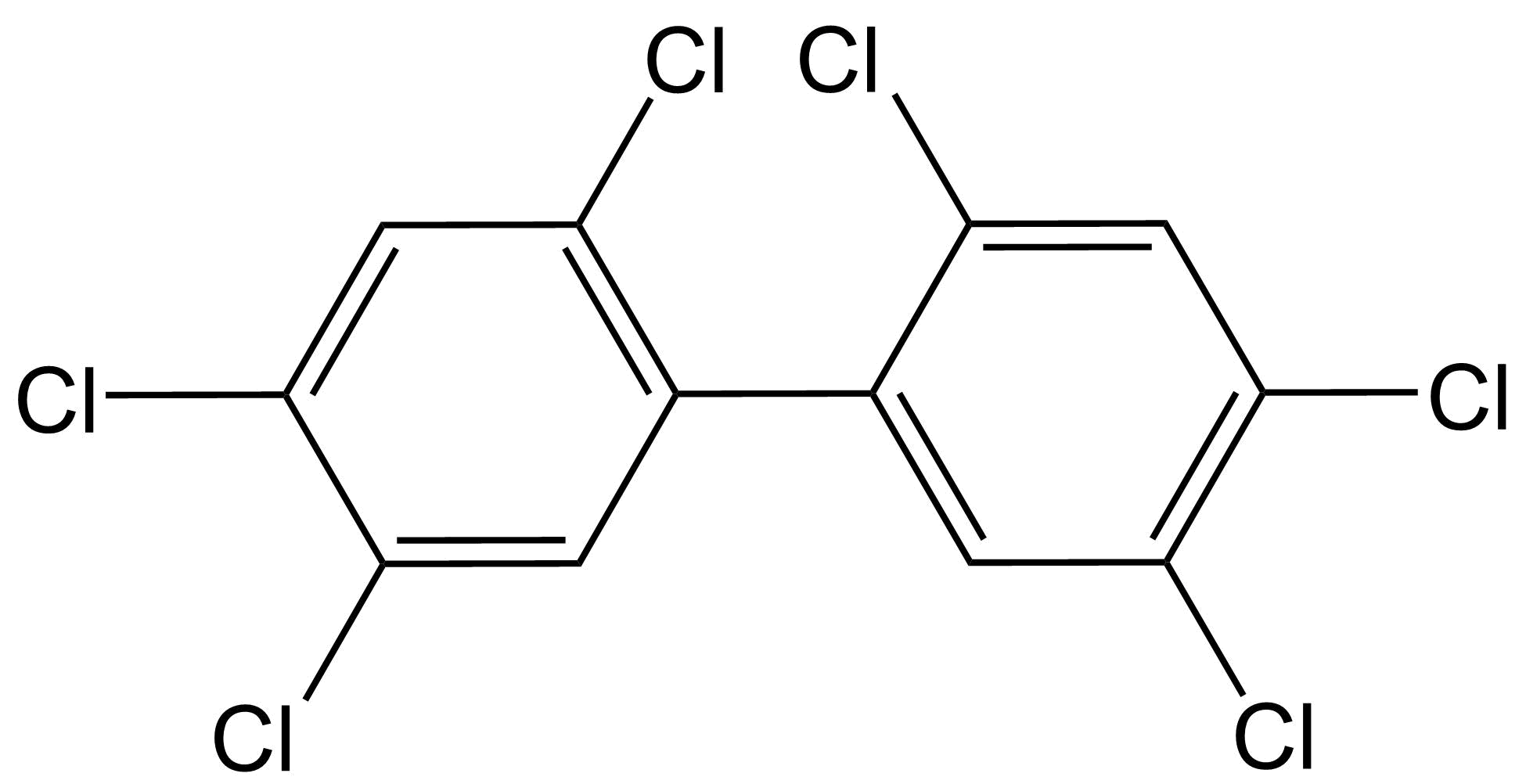
Toxicidad similar a dioxinas El término dioxinas se refiere a compuestos polihalogenados de dibenzo- [p] -dioxina (PHDD), que son moléculas planas que consisten en dos anillos aromáticos halogenados, los cuales están conectados por dos puentes éter. La dioxina más potente y bien estudiada es la 2,3,7,8-tetracloro- [p] -dibenzodioxina (2,3,7,8-TCDD), a la que a menudo se le conoce demasiado simplemente como TCDD o incluso simplemente “dioxina”. Otros compuestos con propiedades similares (compuestos similares a dioxinas) incluyen compuestos de dibenzo- [p] -furano (PHDF) polihalogenados (a menudo denominados “furanos”), que son moléculas planas que consisten en dos anillos aromáticos halogenados conectados por un puente éter y un enlace carbono-carbono. Una tercera clase principal de compuestos similares a las dioxinas pertenece a los bifenilos polihalogenados (PHB), que consisten en dos anillos aromáticos halogenados conectados solo por un enlace carbono-carbono. Los compuestos más conocidos que pertenecen a esta última categoría son los bifenilos policlorados (PCB). De todos los compuestos PHDD, PHDF o PHB, solo los compuestos persistentes y planos se consideran compuestos similares a dioxinas. Para los PHB, esto implica que deben contener cero o como máximo una sustitución halógena en cualquiera de las cuatro posiciones orto (ver ejemplos a continuación). Los PHB no orto-sustituidos pueden obtener fácilmente una confirmación plana con los dos anillos aromáticos en un campo plano, mientras que los PHB mono-orto-sustituidos pueden obtener dicha confirmación a mayores costos energéticos.
La composición plana es necesaria para que los compuestos similares a las dioxinas encajen como llave en la cerradura del receptor de arilhidrocarburo (AhR) (también conocido como el “receptor de dioxinas o DR), presente en el citosol. El AhR activado luego se disocia de sus proteínas represoras, se transloca al núcleo y forma un heterodímero con el translocador nuclear AhR (ARNT). El complejo Ahr-ARNt se une a elementos de respuesta a dioxinas (DRE) en las regiones promotoras de genes sensibles a dioxinas en el ADN, conduciendo finalmente a la transcripción y traducción de estos genes (ver Figura 1 en Denison & Nagy, 2003). Ejemplos famosos de tales genes pertenecen a las familias CYP1, UGT y GST, que son las enzimas metabólicas de Fase I y Fase II cuya activación por el complejo Ahr-ARNt es una respuesta natural desencadenada por la necesidad de eliminar xenobióticos (enlace a la sección sobre Metabolismo y defensa xenobióticos). Otros genes con un DRE en su región promotora incluyen genes involucrados en la fosforilación de proteínas, como el protooncogen c-raf y el inhibidor de quinasa dependiente de ciclina p27. Este mecanismo clásico de inducción de expresión génica dependiente del complejo ligando:AHR:ARNT:Dre, sin embargo, no puede explicar todos los diferentes tipos de toxicidad observados para las dioxinas, incluyendo inmunotoxicidad, toxicidad reproductiva y toxicidad para el desarrollo. Aún así, se sabe que estos efectos también están mediados a través de la AhR, ya que no se observaron en ratones knockout de AhR. Esto puede explicarse en parte por el hecho de que aún no se conocen todos los genes que están bajo control transcripcional de un DRE. Además, se han descrito mecanismos dependientes de AhR distintos de este mecanismo clásico. Por ejemplo, la activación de AhR puede tener efectos antiestrogénicos debido a que AhR activado (1) se une al receptor de estrógeno (ER) y lo dirige para la degradación, (2) se une (con ARNT) a DR inhibidores en el promotor de genes dependientes de ER, y (3) compite con el dímero ER por coactivadores comunes. Aunque los compuestos similares a las dioxinas requieren absolutamente que el AhR ejerza sus principales efectos toxicológicos, también se han descrito varios efectos independientes de AhR, como alteraciones independientes de AHR en la expresión génica y cambios en la afluencia de Ca 2+ relacionados con cambios en la actividad de la proteína quinasa. Aparte de los compuestos similares a dioxinas halogenados persistentes descritos anteriormente, otros compuestos también pueden activar el AhR, incluidos los agonistas naturales de AhR (NaHRas) que se encuentran en los alimentos (por ejemplo, indolo [3,2-b] carbazol (ICZ) en vegetales crucíferos, bergamottina en pomelos, tangeretina en cítricos) y otros productos planos compuestos aromáticos, incluidos los hidrocarburos aromáticos policíclicos (HAP) producidos por la combustión incompleta de combustibles orgánicos. Tras la activación del AhR, estos compuestos no persistentes son metabolizados por las enzimas de biotransformación CYP1A inducidas. Además, se ha identificado un ligando endógeno de AhR llamado 6-formilindolo [3,2-b] carbazol (FICZ). El FICZ es un mediador en muchos procesos fisiológicos, incluyendo las respuestas inmunes, el crecimiento celular y la diferenciación. Los niveles endógenos de FICZ están regulados por un bucle de retroalimentación negativa FICZ/AHR/CYP1a, es decir, FICZ activa AhR y es metabolizado por el CYP1A inducido posteriormente. La desregulación de este bucle de retroalimentación negativa por otros agonistas de AhR puede alterar el funcionamiento de FICZ, y posiblemente podría explicar algunos de los efectos observados para los compuestos similares a las dioxinas. Lectura adicional: Denison, M.S., Soshilov, A.A., He, G., De Groot, D.E., Zhao, B. (2011). Exactamente lo mismo pero diferente: promiscuidad y diversidad en los mecanismos moleculares de acción del Receptor Arilo Hidrocarbonado (Dioxina). Ciencias Toxicológicas 124, 1-22. |
Lectura adicional:
Boelsterli, U.A. (2009). Toxicología Mecánica (2ª edición). Informa Healthcare, Nueva York, Londres.
Le Ferrec, E., Øvrevik J. (2018). Receptores acoplados a proteína G (GPCR) y exposición ambiental. Consecuencias para el metabolismo celular usando los adrenoceptores b como ejemplo. Dictamen Actual en Toxicología 8, 14-19.
cursos.washington.edu/conj/bess/gpcr/gpcr.htm
¿Por qué los compuestos interfieren con los canales iónicos principalmente compuestos neurotóxicos?
La señalización GPRC no solo se interrumpe a través de la interacción con el receptor. ¿Qué mecanismos alternativos pueden desempeñar un papel?
¿Cuál es el principal efecto de activar los receptores ligados a enzimas?
¿Qué sucede si un compuesto se une a un receptor nuclear?
4.2.3. Estrés oxidativo - I.
Especies reactivas de oxígeno y antioxidantes
Autor: Frank van Belleghem
Revisores: Raymond Niesink, Kees van Gestel, Éva Hideg
Objetivos de aprendizaje:
Deberías ser capaz de
- explicar qué es el estrés oxidativo, bajo qué circunstancias surge y por qué es importante en toxicología.
- describir qué especies reactivas de oxígeno son y cómo se producen.
- describir cómo se mantienen bajo control los niveles de especies reactivas de oxígeno.
- hacer una distinción entre antioxidantes enzimáticos y no enzimáticos.
Palabras clave: Especies reactivas de oxígeno, Reacción de Fenton, Antioxidantes enzimáticos, Antioxidantes no enzimáticos
Especies reactivas de oxígeno
El oxígeno molecular (O 2) es un subproducto de la fotosíntesis y esencial para todas las células heterotróficas porque funciona como aceptor terminal de electrones durante la oxidación de sustancias orgánicas en la respiración aeróbica. Este proceso da como resultado la reducción de O 2 a agua, lo que lleva a la formación de energía química y a la reducción de potencia. La razón por la que el O 2 puede reducirse con relativa facilidad en los sistemas biológicos se puede encontrar en las propiedades fisicoquímicas de la molécula de oxígeno (en el estado básico triplete, es decir, como ocurre en la atmósfera). Debido a su configuración electrónica, O 2 es en realidad un birradical que puede actuar como aceptor de electrones. Los orbitales moleculares externos de O 2 contienen cada uno un electrón, los espines de estos electrones son paralelos (Figura 1). En consecuencia, el oxígeno (en estado fundamental) no es muy reactivo porque, según el principio de exclusión de Pauli, solo un electrón a la vez puede reaccionar con otros electrones en un enlace covalente. Como consecuencia, el oxígeno solo puede sufrir reducciones univalentes, y la reducción completa del oxígeno al agua requiere la adición secuencial de cuatro electrones que conducen a la formación de intermedios de oxígeno de uno, dos, tres electrones (Figura 1). Estos intermedios de oxígeno son, en secuencia, el radical anión superóxido (O 2 ●-), el peróxido de hidrógeno (H 2 O 2) y el radical hidroxilo (● OH).
Otra especie reactiva de oxígeno de importancia es el oxígeno singlete (1 O 2 o 1 Δ g). El oxígeno singlete se forma convirtiendo el oxígeno molecular en estado fundamental en un estado de energía excitada, que es mucho más reactivo que el oxígeno molecular normal en estado fundamental. El oxígeno singlete se genera típicamente mediante un proceso llamado fotosensibilización, por ejemplo en el cristalino del ojo. La fotosensibilización ocurre cuando la absorción de luz (UV) por una sustancia endógena o xenobiótica eleva el compuesto a un estado de mayor energía (un intermedio triplete de alta energía) que puede transferir su energía al oxígeno, formando oxígeno singlete altamente reactivo. Además de las reacciones fotodinámicas dependientes de oxígeno, el oxígeno singlete también es producido por los neutrófilos y se ha sugerido que esto es importante para la destrucción bacteriana a través de la formación de ozono (O 3) (Onyango, 2016).
Debido a que estos intermedios de oxígeno son productos potencialmente nocivos que pueden dañar los componentes celulares, se les conoce como especies reactivas de oxígeno (ROS). Las ROS también se denominan a menudo 'radicales libres' pero esto es incorrecto porque no todas las ROS son radicales (por ejemplo, H 2 O 2, 1 O 2 y O 3). Además, como todos los radicales son (actualmente) considerados como no unidos, el prefijo 'libre' en realidad es innecesario (Koppenol y Traynham, 1996).
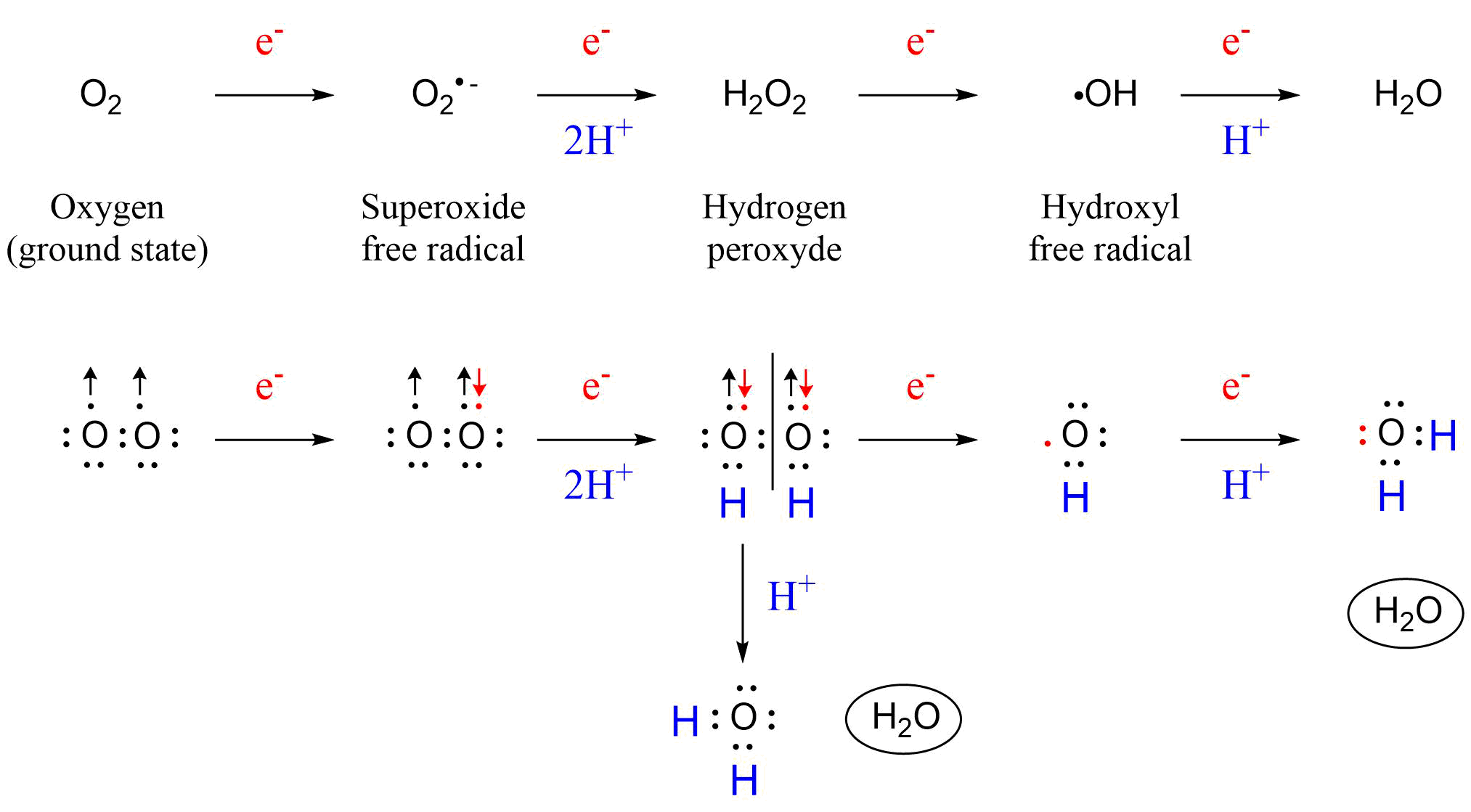
Las ROS son subproductos del metabolismo aeróbico en los diferentes orgánulos de las células, por ejemplo la respiración o la fotosíntesis, o como parte de las defensas contra patógenos. Las fuentes endógenas de especies reactivas de oxígeno incluyen la fosforilación oxidativa, el metabolismo de P450, los peroxisomas y la activación celular inflamatoria. Por ejemplo, los radicales aniónicos superóxido se forman endógenamente a partir de la reducción de oxígeno por la semiquinona de ubiquinona (coenzima Q), una coenzima ampliamente distribuida en plantas, animales y microorganismos. Las ubiquinonas funcionan junto con enzimas en la respiración celular (es decir, procesos de oxidación-reducción). El radical anión superóxido se forma cuando un electrón es absorbido por uno de los orbitales π*-antienlaces (formados por dos orbitales atómicos 2p) de oxígeno molecular.
Un segundo ejemplo de una fuente endógena de radicales aniónicos superóxido es la auto-oxidación de proteínas hemo reducidas. Se sabe, por ejemplo, que los complejos de sustrato de oxiferrocitocromo P-450 pueden sufrir autooxidación y posteriormente dividirse en (ferri) citocromo P-450, un radical anión superóxido y el sustrato (S). Este proceso se conoce como el desacoplamiento del ciclo del citocromo P-450 (CYP) y también se conoce como la actividad oxidasa del citocromo P-450. No obstante, cabe mencionar que este no es el funcionamiento normal del CYP. Sólo cuando la transferencia de un átomo de oxígeno a un sustrato no está estrechamente acoplada a la utilización del NADPH, de manera que los electrones derivados del NADPH se transfieren al oxígeno para producir O 2 ●- (y también H 2 O 2).
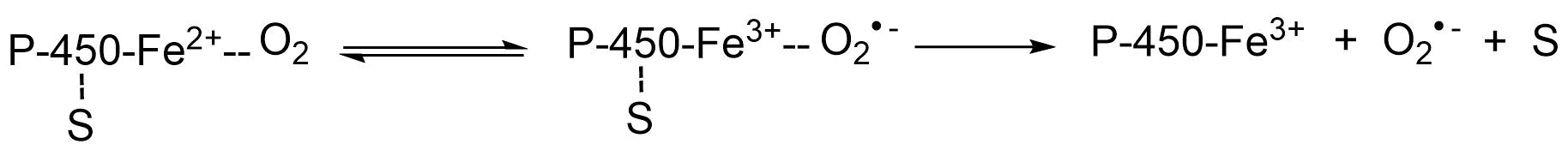
En el Cuadro 1 se muestran las especies clave de oxígeno y su vida media biológica, su distancia de migración, la fuente endógena y su reacción con compuestos biológicos.
Cuadro 1. Las especies clave de oxígeno y sus características (tabla adaptada de Das & Roychoudhury, 2014)
|
Especies de ROS |
Vida media (T 1/2) |
Distancia de migración |
Fuente endógena |
Modo de acción |
|
Radical anión superóxido (O 2 ●-) |
1-4 µs |
30 nm |
Mitocondrias, citocromo P450, macrófagos/ células inflamatorias, membranas, cloroplastos |
Reacciona con compuestos con dobles enlaces |
|
Radical hidroxilo (● OH) |
1 µs |
1 nm |
Mitocondrias, membranas, cloroplastos |
Reacciona vigorosamente con todas las biomoléculas. |
|
Peróxido de hidrógeno (H 2 O 2) |
1 ms |
1 µm |
Mitocondrias, membranas, peroxisomas, cloroplastos |
Oxida las proteínas al reaccionar con el residuo de Cys. |
|
Oxígeno Singlete |
1-4 µs |
30 nm |
Mitocondrias, membranas, cloroplastos |
Oxida proteínas, ácidos grasos poliinsaturados y ADN |
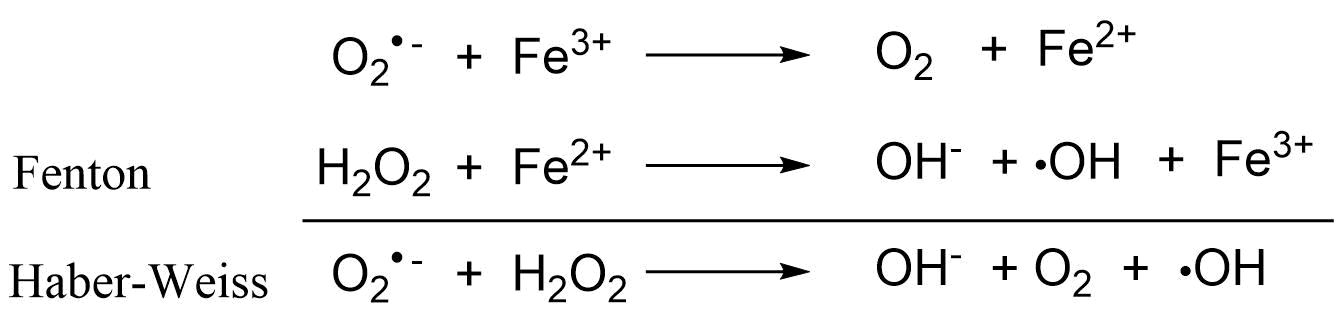
Debido a su reactividad, a niveles elevados, las ROS pueden dañar indiscriminadamente componentes celulares como lípidos, proteínas y ácidos nucleicos. En particular, el radical anión superóxido y los radicales hidroxilo que poseen un electrón desapareado son muy reactivos. De hecho, el hidroxilo tiene el mayor potencial de reducción de 1 electrón, lo que lo convierte en el radical más reactivo conocido. Los radicales hidroxilo (Figura 1) pueden surgir del peróxido de hidrógeno en presencia de metal de transición redox activo, notablemente Fe 2+/3+ o Cu +/2+, a través de la reacción de Fenton. En el caso del hierro, para que esta reacción tenga lugar, la forma oxidada (Fe 3+) tiene que reducirse a Fe 2+. Esto significa que el Fe 2+ solo se libera en un ambiente ácido (hipoxia local) o en presencia de radicales aniónicos superóxido. La reducción de Fe 3+, seguida de la interacción con el peróxido de hidrógeno, que conduce a la generación del radical hidroxilo, se denomina reacción de Haber-Weiss catalizada por hierro.

Mantener bajo control las especies reactivas de oxígeno
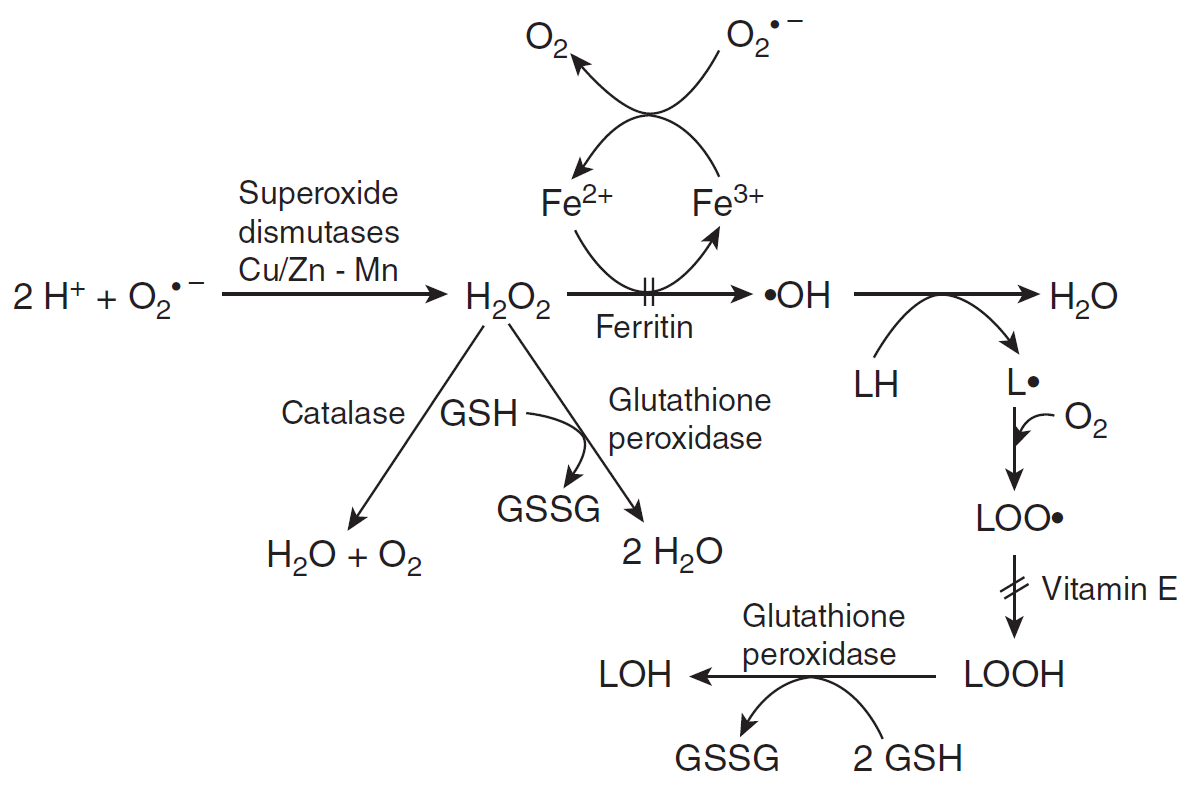
Para mantener las concentraciones de ROS en niveles fisiológicos bajos, los organismos aeróbicos han desarrollado complejos sistemas de defensa antioxidante que incluyen componentes antioxidantes enzimáticos y no enzimáticos. Estos son mecanismos celulares que se desarrollan para inhibir la oxidación mediante la extinción de ROS. Se sabe que tres clases de enzimas proporcionan protección contra especies reactivas de oxígeno: las superóxido dismutasas que catalizan la dismutación del radical anión superóxido, y las catalasas y peroxidasas que reaccionan específicamente con peróxido de hidrógeno. Estas enzimas antioxidantes pueden verse como una defensa de primera línea ya que impiden la conversión de las especies de oxígeno menos reactivas, el radical anión superóxido y el peróxido de hidrógeno, a especies más reactivas como el radical hidroxilo. La segunda línea de defensa consiste en gran parte en sustancias no enzimáticas que eliminan radicales como el glutatión y las vitaminas E y C. En la Figura 3 se proporciona una visión general del sistema de defensa celular.
Antioxidantes enzimáticos
Las superóxido dismutasas (SOD) son proteínas que contienen metales (metaloenzimas) que catalizan la dismutación del radical anión superóxido a oxígeno molecular en estado fundamental y peróxido de hidrógeno, como se ilustra mediante las siguientes reacciones:
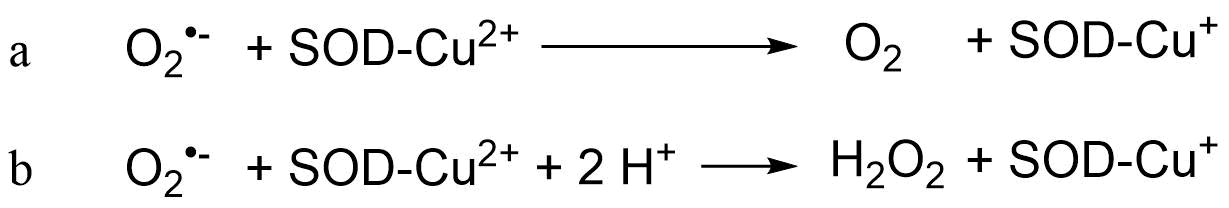
La dismutación de los radicales aniónicos superóxido actúa en la primera parte de la reacción con el radical anión superóxido como agente reductor (a), y como oxidante en la segunda parte (b). Diferentes tipos de SOD se localizan en diferentes localizaciones celulares, por ejemplo Cu-Zn-SOD se localizan principalmente en el citosol de eucariotas, Mn-SOD en mitocondrias y procariotas, Fe-SOD en cloroplastos y procariotas y Ni-SOD en procariotas. Mn, Fe, Cu y Ni son los metales activos redox en las enzimas, mientras que el Zn no es catalítico en el Cu-Zn-SOD.
H 2 O 2 se degrada adicionalmente por catalasa y peroxidasa. La catalasa (CAT) contiene cuatro grupos hemo que contienen hierro que permiten que la enzima reaccione con el peróxido de hidrógeno y generalmente se localiza en peroxisomas, que son orgánulos con una alta tasa de producción de ROS. La catalasa convierte el peróxido de hidrógeno en agua y oxígeno. De hecho, la catalasa coopera con la superóxido dismutasa en la eliminación del peróxido de hidrógeno resultante de la reacción de dismutación. La catalasa actúa solo sobre el peróxido de hidrógeno, no sobre el hidroperóxido orgánico.
Las peroxidasas (Px) son hemoproteínas que utilizan H 2 O 2 para oxidar una variedad de sustratos endógenos y exógenos. Una importante familia de enzimas peroxidasas es la glutatión peroxidasa (GPx) que contiene selenio-cisteína, presente en el citosol y mitocondrias. Cataliza la conversión de hidrógeno H 2 O 2 a H 2 O mediante la oxidación del glutatión reducido (GSH) en su forma disulfuro de glutatión (GSSG). La glutatión peroxidasa cataliza no sólo la conversión del peróxido de hidrógeno, sino también la de los peróxidos orgánicos. Puede transformar diversos peróxidos, por ejemplo los hidroperóxidos de lípidos. La glutatión peroxidasa se encuentra tanto en el citosol como en las mitocondrias. En el citosol, la enzima está presente en vesículas especiales.
Otro grupo de enzimas, no descritas más aquí, son las Peroxiredoxinas (Prxs), presentes en el citosol, mitocondrias y retículo endoplásmico, utilizan un par de residuos de cisteína para reducir y con ello desintoxicar el peróxido de hidrógeno y otros peróxidos. Hay que mencionar que ninguna enzima reacciona con el radical hidroxilo ni el oxígeno singlete.
Antioxidantes no enzimáticos
La segunda línea de defensa consiste en gran parte en sustancias no enzimáticas que eliminan los radicales. El principal antioxidante es el glutatión (GSH), que actúa como un captador nucleófilo de compuestos tóxicos, atrapando metabolitos electrófilos formando un enlace tioéter entre el residuo de cisteína de GSH y el electrófilo. El resultado generalmente es un conjugado menos reactivo y más soluble en agua que puede excretarse fácilmente (ver también reacciones de biotransformación de fase II). GSH también es un cosustrato para la degradación enzimática (catalizada por peroxidasa GS) de H 2 O 2 y mantiene las células en un estado reducido y participa en la regeneración de proteínas oxidadas.
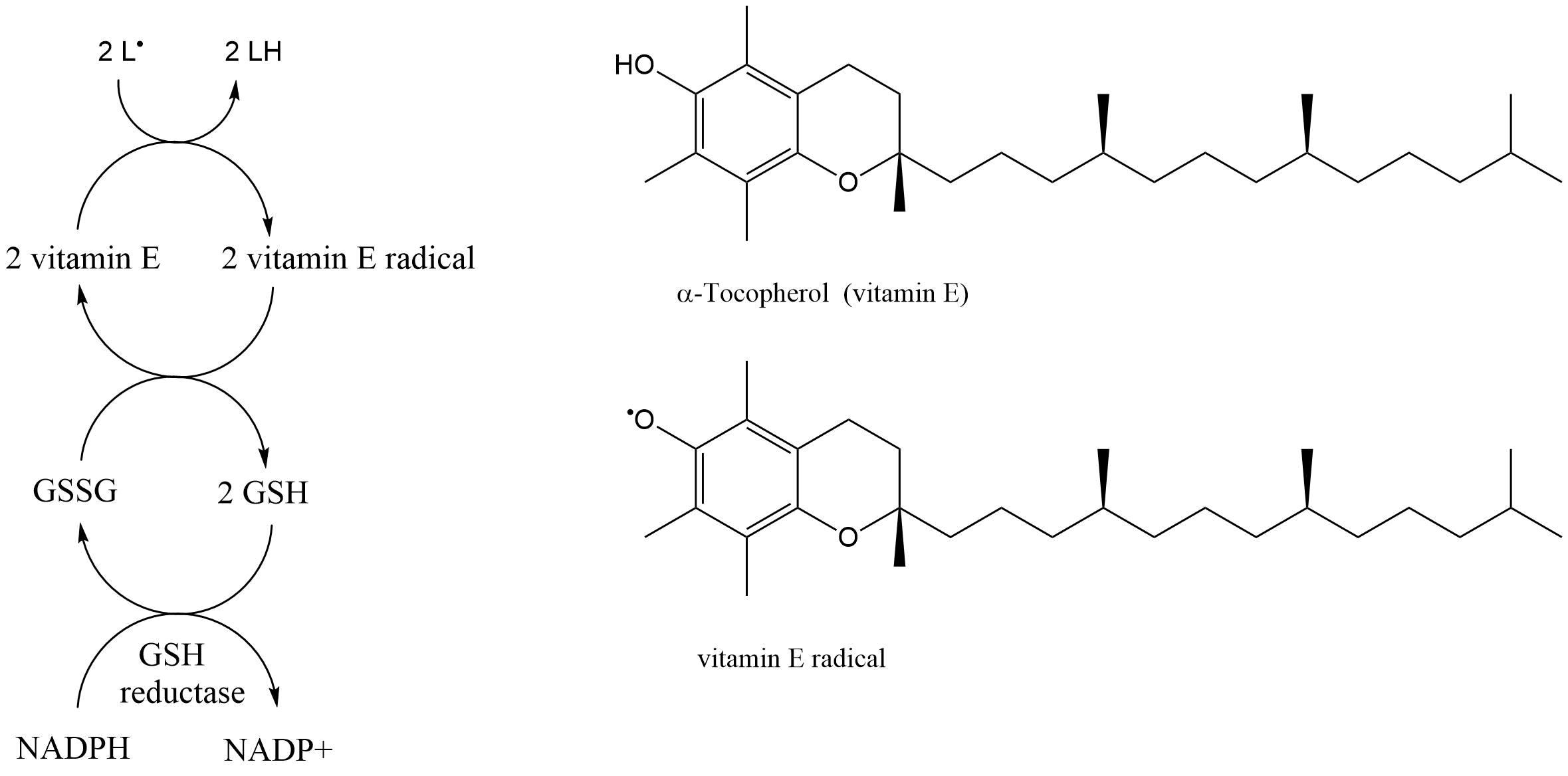
Otros eliminadores radicales importantes de la célula son las vitaminas E y C. La vitamina E (α-tocoferol) es lipofílica y se incorpora en las membranas celulares y orgánulos subcelulares (retículo endoplásmico, mitocondrias, núcleos celulares) y reacciona con peróxidos lipídicos. α-tocoferol puede ser dividido en dos partes, una cola de fitilo lipófila (intercalada con residuos de ácidos grasos de fosfolípidos) y una cabeza de cromano más hidrófila con un grupo fenólico (frente al citoplasma). Este grupo fenólico puede reducir radicales (por ejemplo, radicales peroxi lipídicos (LOO ●, ver Figura 2, para la explicación de la peroxidación lipídica, ver sección Estrés oxidativo II: inducción por exposición química y posibles efectos) y por lo tanto se oxida a su vez al radical tocoferilo que es relativamente poco reactivo porque se estabiliza por resonancia. El radical es regenerado por vitamina C o por glutatión reducido (Figura 4). Los antioxidantes oxidados no enzimáticos son regenerados por diversas enzimas como el glutatión.
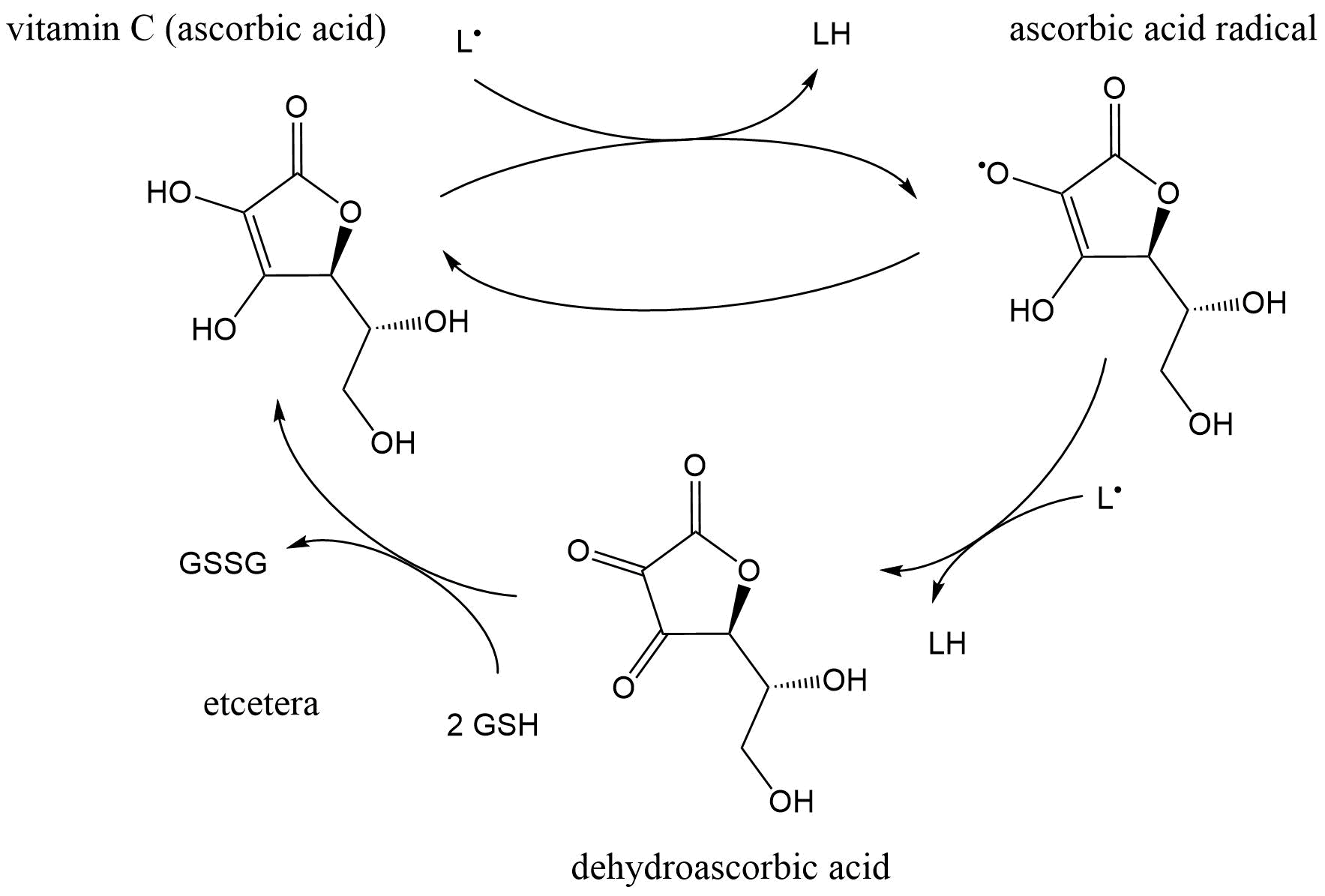
La vitamina C (ácido ascórbico) es un antioxidante soluble en agua y está presente en el citoplasma. El ácido ascórbico es un donante de electrones que reacciona con bastante rapidez con el radical anión superóxido y los radicales peroxilo, pero generalmente es ineficaz para desintoxicar los radicales hidroxilo debido a su extrema reactividad no alcanza el antioxidante (Ver Klaassen, 2013). Además, regenera α-tocoferol en combinación con GSH reducida o compuestos capaces de donar equivalentes reductores (Nimse y Pal, 2015): Figura 5.
Referencias
Bolton, J.L., Dunlap, T. (2016). Formación y dianas biológicas de quinonas: efectos citotóxicos versus citoprotectores. Investigación Química en Toxicología 30, 13-37.
Das, K., Roychoudhury, A. (2014). Especies reactivas de oxígeno (ROS) y respuesta de antioxidantes como captadores de ROS durante el estrés ambiental en plantas. Fronteras en la Ciencia Ambiental 2, 53.
Edreva, A. (2005). Generación y captación de especies reactivas de oxígeno en cloroplastos: un enfoque submolecular.Agricultura, Ecosistemas y Medio Ambiente 106, 119-133.
Klaassen, C. D. (2013). Toxicología de Casarett & Doull: La ciencia básica de los venenos, Octava Edición, McGraw-Hill Professional.
Koppenol, W.H., Traynham, J.G. (1996). Di NO al óxido nítrico: nomenclatura para compuestos que contienen nitrógeno y oxígeno. En: Métodos en Enzimología (Vol. 268, pp. 3-7). Prensa Académica.
Louise Bolton, J. (2014). Vía de bioactivación de quinona metida: ¿contribución a la toxicidad y/o citoprotección?. Química Orgánica Actual 18, 61-69.
Nimse, S.B., Pal, D. (2015). Radicales libres, antioxidantes naturales y sus mecanismos de reacción. Rsc Avances 5, 27986-28006.
Onyango, A.N. (2016). Generación endógena de oxígeno singlete y ozono en tejidos humanos y animales: mecanismos, significación biológica e influencia de componentes dietéticos. Medicina oxidativa y longevidad celular, 2016.
Niesink, R.J.M., De Vries, J., Hollinger, M.A. (1996). Toxicología: Principios y Aplicaciones. Prensa CRC.
Smart, R.C., Hodgson, E. (Eds.). (2018). Toxicología Molecular y Bioquímica. John Wiley & Hijos.
¿Cuáles son las posibilidades de que se formen radicales hidroxilo dentro de la célula? ¿De qué factores depende tal formación?
Dado:
Dos especies de oxígeno:
I oxígeno atmosférico (O 2)
II oxígeno singlete (1 O 2)
¿Qué especies de oxígeno contienen uno o más electrones desapareados y, por lo tanto, tienen propiedades radicales?
I y II
solo yo
solo II
ni I ni II
¿Cuáles de los siguientes radicales son desintoxicados por a-tocoferol (vitamina E)?
I radical hidroxilo, • OH
II radical anión superóxido, O 2 -•
III radical lipídico (L •)
IV radical peroxilo lipídico (LOO •)
I y II
III y IV
I, II y III
II, III y IV
Dado:
Tres enzimas:
I catalasa
II peroxidasa
III superóxido dismutasa
¿Qué enzima elimina el peróxido de hidrógeno?
I y II
I y III
II y III
sólo III
4.2.3. Estrés oxidativo - II.
Inducción por exposición química y posibles efectos
Autor: Frank van Belleghem
Revisores: Raymond Niesink, Kees van Gestel, Éva Hideg
Objetivos de aprendizaje:
Deberías ser capaz de
- explicar cómo los compuestos xenobióticos pueden conducir a una mayor producción de.
- explicar lo que hace el estrés oxidativo con
- proteínas,
- lípidos,
- ADN y
- regulación génica.
Palabras clave: equilibrio prooxidante-antioxidante, bioactivación, daño oxidativo,
Cómo los compuestos xenobióticos inducen la generación de ROS
La formación de especies reactivas de oxígeno (ROS; ver sección Estrés oxidativo I) puede involucrar sustancias endógenas y procesos químico-fisiológicos así como xenobióticos. La evidencia experimental ha demostrado que el estrés oxidativo puede considerarse como uno de los mecanismos clave que contribuyen al daño celular de muchos tóxicos. El estrés oxidativo se ha definido como “una alteración en el equilibrio prooxidante-antioxidante a favor del primero”, lo que lleva a un daño potencial. Es el punto en el que la producción de ROS excede la capacidad de los antioxidantes para prevenir daños (Klaassen et al., 2013).
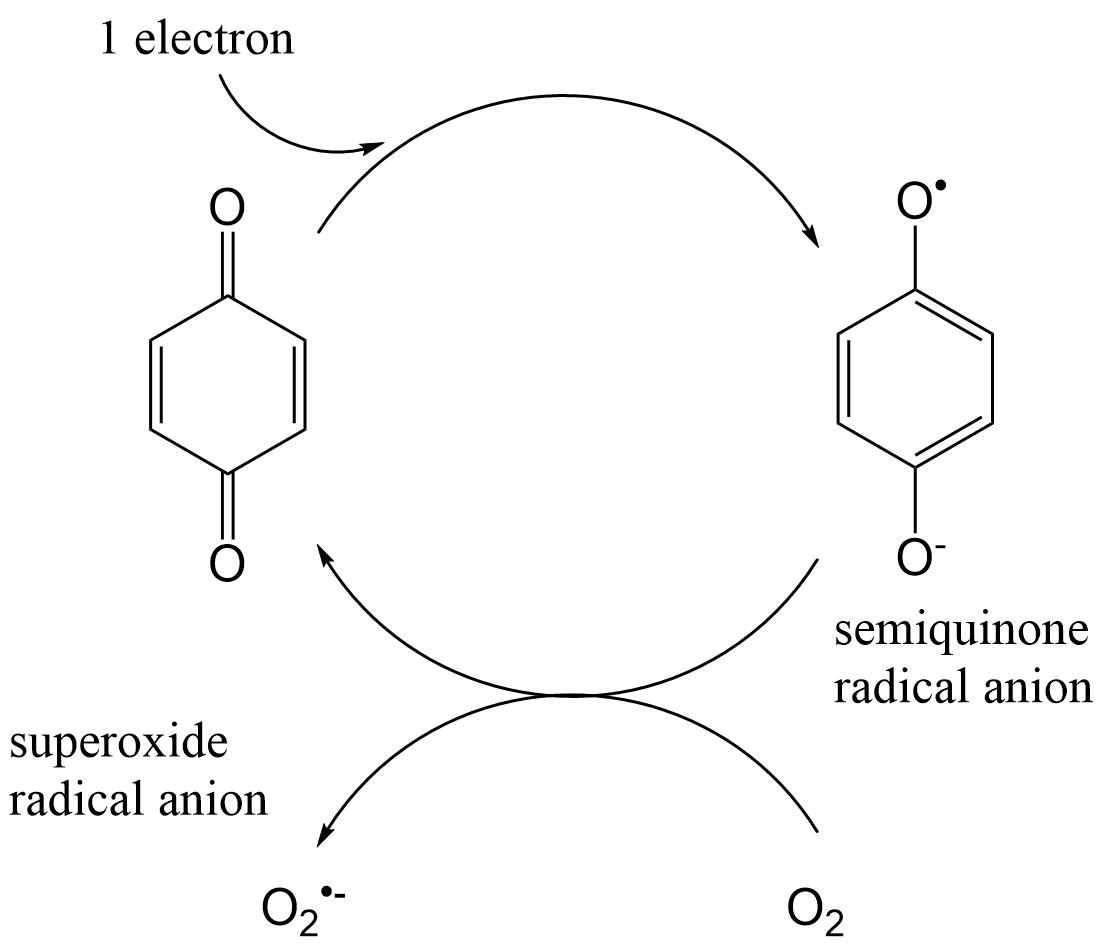
Los xenobióticos involucrados en la formación del radical anión superóxido son principalmente sustancias que pueden ser absorbidas en especies de oxígeno tan reactivas, llamadas ciclos redox. Estas incluyen las quinonas y las hidroquinonas en particular. En el caso de las quinonas el ciclo redox inicia con una etapa de reducción de un electrón, igual que en el caso de la benzoquinona (Figura 1). Posteriormente, la benzosemiquinona resultante pasa el electrón recibido al oxígeno molecular. La reducción de quinonas es catalizada por la citocromo P-450 reductasa dependiente de NADPH.
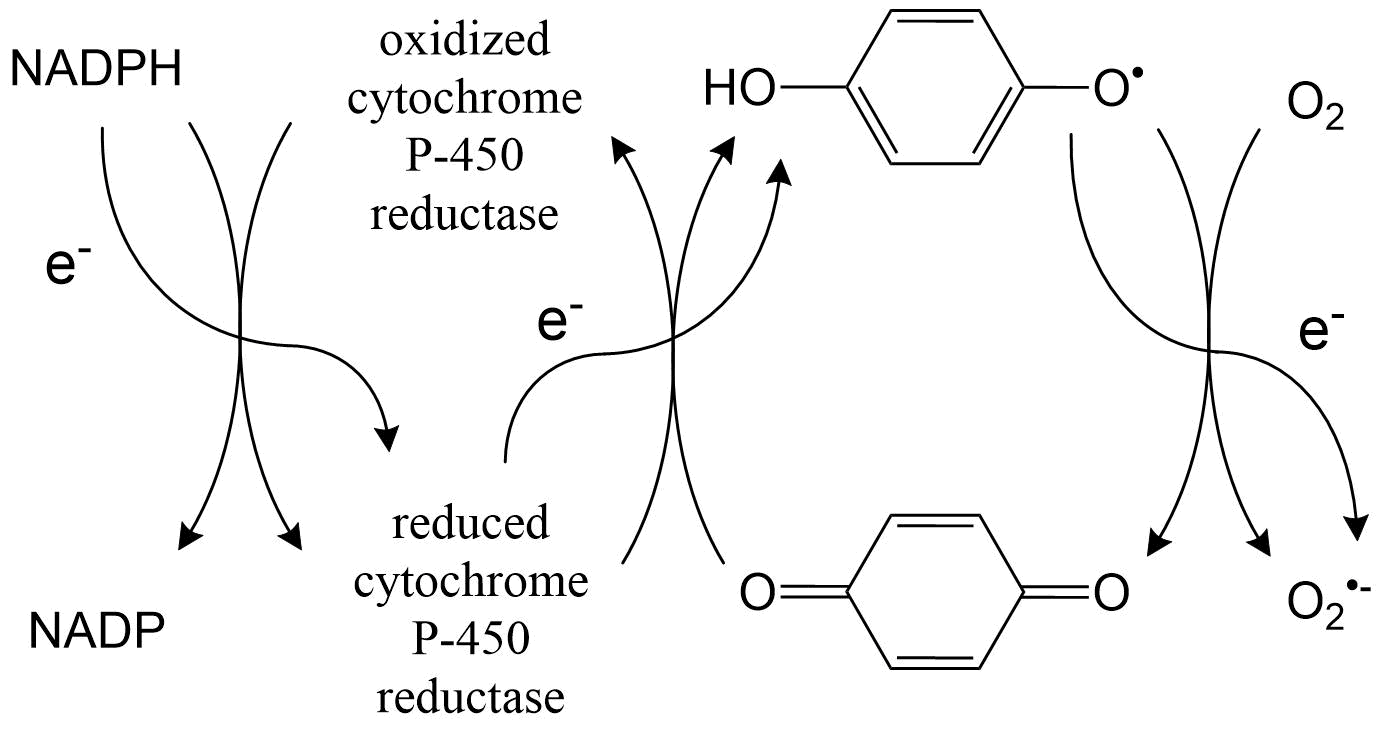
Obviamente, las hidroquinonas pueden ingresar a un ciclo redox a través de un paso oxidativo. Esta etapa puede ser catalizada por enzimas, por ejemplo prostaglandina sintasa.
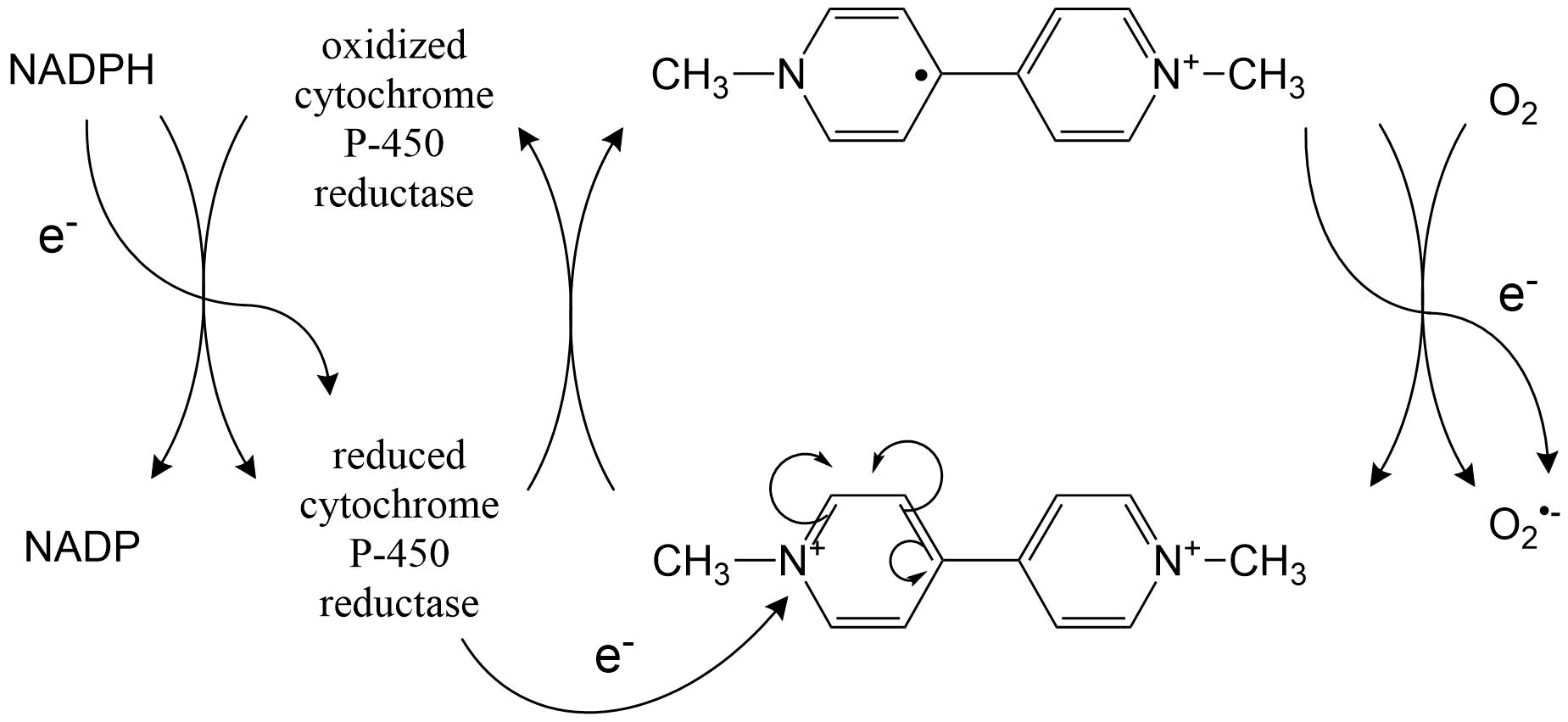
Otros tipos de xenobióticos que pueden ser absorbidos en un ciclo redox, son los derivados bipiridilo. Un ejemplo bien conocido es el herbicida paraquat, que causa lesiones al tejido pulmonar en humanos y animales. La Figura 2 muestra esquemáticamente su bioactivación. Otros compuestos que son absorbidos en un ciclo redox son nitroaromáticos, compuestos azo, hidroxilaminas aromáticas y ciertos quelatos metálicos (particularmente Cu y Zn).
Los xenobióticos pueden mejorar la producción de ROS si son capaces de ingresar a mitocondrias, microsomas o cloroplastos e interactuar con las cadenas de transporte de electrones, bloqueando así el flujo normal de electrones. Como consecuencia, y especialmente si los compuestos son aceptores de electrones, desvían el flujo normal de electrones e incrementan la producción de ROS. Un ejemplo típico es el fármaco citostático doxorrubicina, un conocido agente quimioterapéutico, que se utiliza en el tratamiento de una amplia variedad de cánceres. La doxorrubicina tiene una alta afinidad por la cardiolipina, un compuesto importante de la membrana mitocondrial interna y por lo tanto se acumula en esa ubicación subcelular.
Los xenobióticos pueden causar daño oxidativo indirectamente al interferir con los mecanismos antioxidantes. Por ejemplo se ha sugerido que como metal no Fenton, el cadmio (Cd) es incapaz de inducir directamente ROS. Sin embargo, indirectamente, Cd induce estrés oxidativo mediante un desplazamiento de metales activos redox, agotamiento de depuradores redox (glutatión) e inhibición de enzimas antioxidantes (grupos sulfhidrilo unidos a proteínas) (Cuypers et al., 2010; Thévenod et al., 2009).
Los mecanismos del estrés oxidativo
Como se mencionó anteriormente, el estrés oxidativo se ha definido como “una alteración en el equilibrio prooxidante-antioxidante a favor del primero”. Las ROS pueden dañar proteínas, lípidos y ADN a través de la oxidación directa, o a través de sensores redox que transducen señales, que a su vez pueden activar procesos que dañan las células como la apoptosis.
Daño proteico oxidativo
La generación de ROS inducida por xenobióticos puede dañar proteínas a través de la oxidación de cadenas laterales de residuos de aminoácidos, la formación de reticulaciones proteína-proteína y la fragmentación de proteínas debido a la oxidación de la cadena principal peptídica. Los aminoácidos cisteína y metionina que contienen azufre son particularmente susceptibles a la oxidación. Un ejemplo de oxidación de cadena lateral es la interacción directa del radical anión superóxido con grupos sulfhidrilo (tiol), formando así radicales tiilo como intermedios:
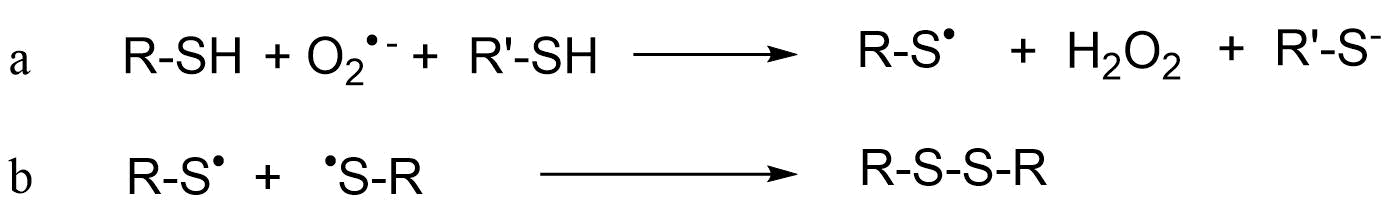
Como consecuencia, el glutatión, compuesto por tres aminoácidos (cisteína, glicina y glutamato) y un importante agente reductor celular, puede dañarse de esta manera. Esto significa que si la oxidación no puede ser compensada o reparada, el estrés oxidativo puede llevar al agotamiento de los equivalentes reductores, lo que puede tener efectos perjudiciales en la célula.
Afortunadamente, los mecanismos de defensa antioxidante limitan el estrés oxidativo y la célula tiene mecanismos de reparación para revertir el daño. Por ejemplo, las proteínas de choque térmico (hsp) son capaces de renaturalizar las proteínas dañadas y las proteínas dañadas oxidativamente son degradadas por el proteasoma.
Daño lipídico oxidativo El
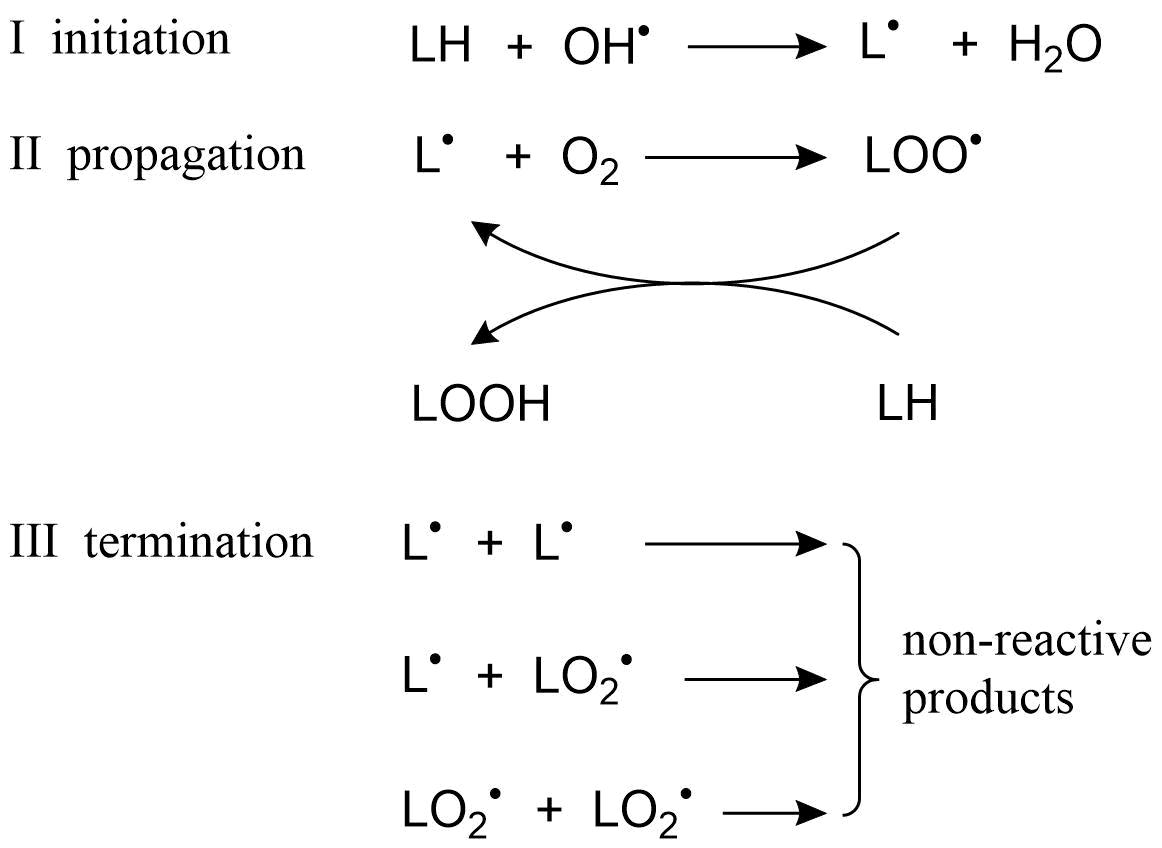
aumento de las concentraciones de radicales reactivos de oxígeno puede causar daño a la membrana debido a la peroxidación lipídica (oxidación de lípidos poliinsaturados) Este daño puede resultar en una fluidez de membrana alterada, actividad enzimática y permeabilidad de la membrana y características de transporte. Una característica importante que caracteriza la peroxidación lipídica es el hecho de que el daño inicial inducido por radicales en un determinado sitio en un lípido de membrana se amplifica fácilmente y se propaga de manera similar a una reacción en cadena, dispersando así el daño a través de la membrana celular. Además, los productos que surgen de la peroxidación lipídica (por ejemplo, radicales alcoxi o aldehídos tóxicos) pueden ser igualmente reactivos que las propias ROS originales y dañar las células por mecanismos adicionales. La reacción en cadena de la peroxidación lipídica consta de tres etapas:
- Abstracción de un átomo de hidrógeno de una cadena de ácidos grasos poliinsaturados por radicales reactivos de oxígeno (formación de radicales, iniciación).
- Reacción del radical ácido graso resultante con oxígeno molecular (oxigenación o, más específicamente, peroxidación, propagación)
- Estos eventos pueden ir seguidos de un proceso de desintoxicación, en el que se detiene la cadena de reacción. Este proceso, que puede proceder en varios pasos, a veces se denomina terminación.
La Figura 3 resume las diversas etapas en la peroxidación lipídica.
En la etapa II, la peroxidación de biomembranas genera una variedad de electrófilos reactivos como epóxidos (LOO •) y aldehídos, incluyendo malondialdehído (MDA). MDA es un aldehído altamente reactivo que exhibe reactividad hacia nucleófilos y puede formar dímeros MDA-MDA. Tanto los dímeros MDA como MDA-MDA son mutagénicos e indicativos del daño oxidativo de los lípidos de una variedad de tóxicos.
Un ejemplo clásico de bioactivación xenobiótica a un radical libre que inicia la peroxidación lipídica es la conversión dependiente del citocromo P450 de tetracloruro de carbono (CCl 4) para generar el radical triclorometilo (• CCl 3) y luego el radical peroxilo triclorometilo CCl 3 OO •. También la citotoxicidad del hierro libre se atribuye a su función como donante de electrones para la reacción de Fenton (ver sección sobre Estrés oxidativo I) por ejemplo a través de la generación de radicales aniónicos superóxido por ciclo redox del paraquat) que conduce a la formación del radical hidroxilo altamente reactivo, un conocido iniciador de la peroxidación lipídica.
Daño oxidativo del ADN Las
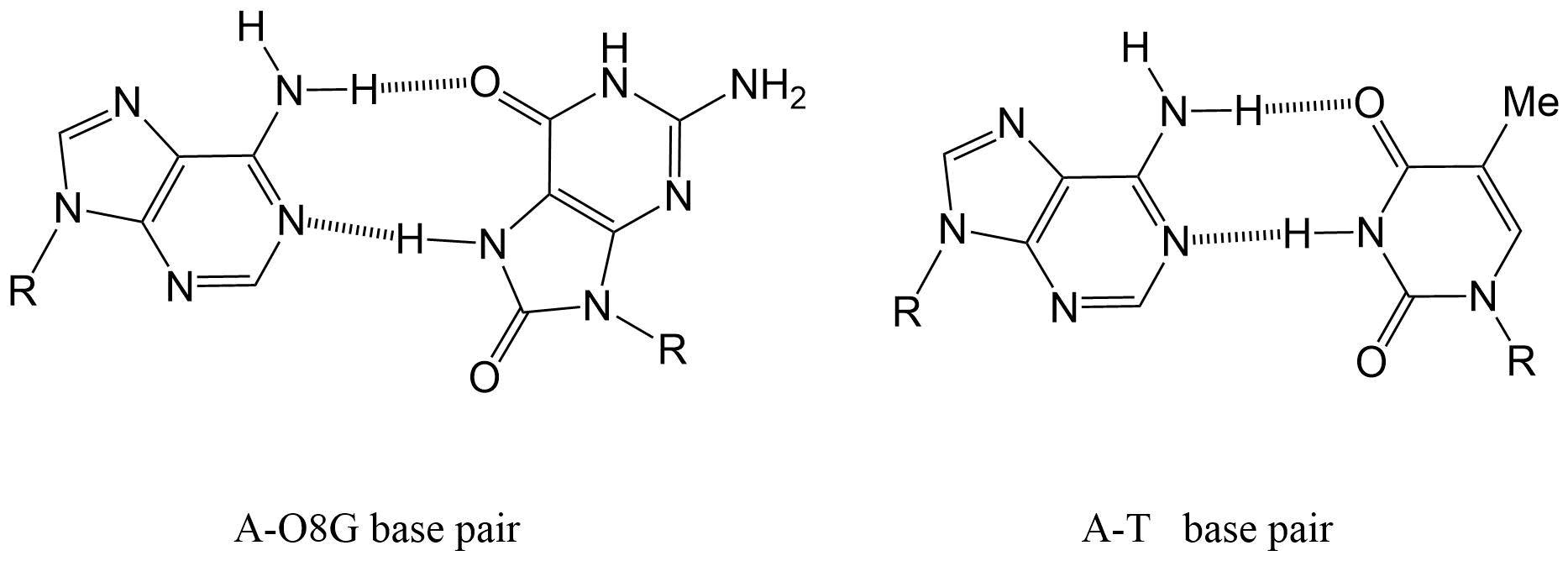
ROS también pueden oxidar bases de ADN y azúcares, producir roturas de ADN monocatenarias o bicatenarias, modificaciones de purina, pirimidina o desoxirribosa y reticulaciones de ADN. Una modificación común al ADN es la hidroxilación de bases de ADN que conduce a la formación de aductos de ADN oxidados. Aunque estos aductos han sido identificados en las cuatro bases de ADN, la guanina es la más susceptible al daño oxidativo porque tiene el menor potencial de oxidación de todas las bases de ADN. La oxidación de guanina y por radicales hidroxilo conduce a la formación de 8-hidroxiguanosina (8-OH-dG) (Figura 4).
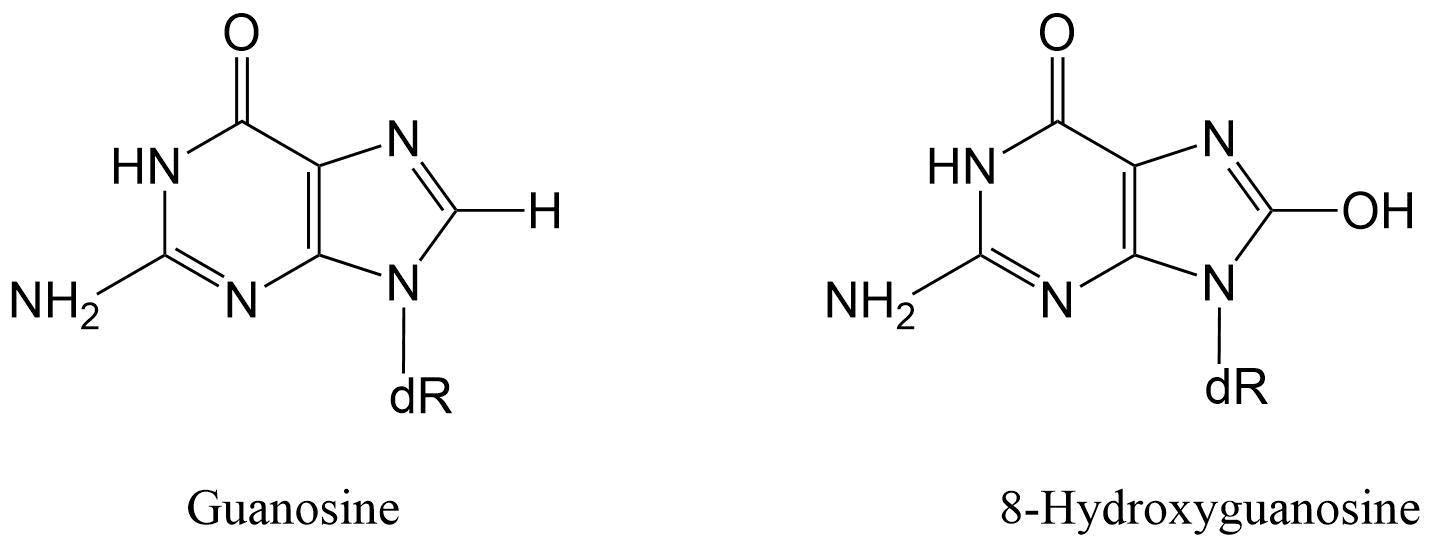
La oxidación de la guanina tiene un efecto perjudicial sobre el pelado de bases, ya que en lugar de enlaces de hidrógeno con citosina como lo hace normalmente la guanina, puede formar un par de bases con adenina. Como resultado, durante la replicación del ADN, la ADN polimerasa puede insertar erróneamente una adenosina opuesta a una 8-oxo-2'-desoxiguanosina (8-oxo-dG), dando como resultado un cambio estable en la secuencia de ADN, proceso conocido como mutagénesis (Figura 5).
Afortunadamente, existe un mecanismo de reparación extenso que mantiene las mutaciones a un nivel relativamente bajo. Sin embargo, el daño persistente del ADN puede dar como resultado errores de replicación, inducción o inhibición de la transcripción, inducción de vías de transducción de señales e inestabilidad genómica, eventos que posiblemente están involucrados en la carcinogénesis (Figura 6). Hay que mencionar que el ADN mitocondrial, es más susceptible al daño oxidativo de bases en comparación con el ADN nuclear debido a su proximidad a la cadena de transporte de electrones (fuente de ROS), y el hecho de que el ADN mitocondrial no está protegido por histonas y tiene un sistema limitado de reparación del ADN.
Figura en preparación
Figura 6. Daño oxidativo por ROS que conduce a mutaciones y eventualmente a la formación de tumores. Figura adaptada de Boelsterli (2002).
Un grupo de xenobióticos que se han asociado claramente con provocar daño oxidativo en el ADN y cáncer son los metales redox activos, incluyendo Fe (III), Cu (II), Ag (I), Cr (III), Cr (VI), que pueden implicar, como se vio anteriormente, la producción de radicales hidroxilo. Otros metales (no redox activos) que pueden inducir la formación de ROS ellos mismos o participar en las reacciones que conducen a ROS generados endógenamente son Pb (II), Cd (II), Zn (II) y el metaloide As (III) y As (V). Compuestos como los hidrocarburos aromáticos policíclicos (HAP), probablemente la familia más grande de contaminantes con efectos genotóxicos, requieren activación por metabolismo endógeno para volverse reactivos y capaces de modificar el ADN. Esta activación es provocada por la denominada biotransformación de Fase I (ver Sección Metabolismo y defensa xenobióticos).
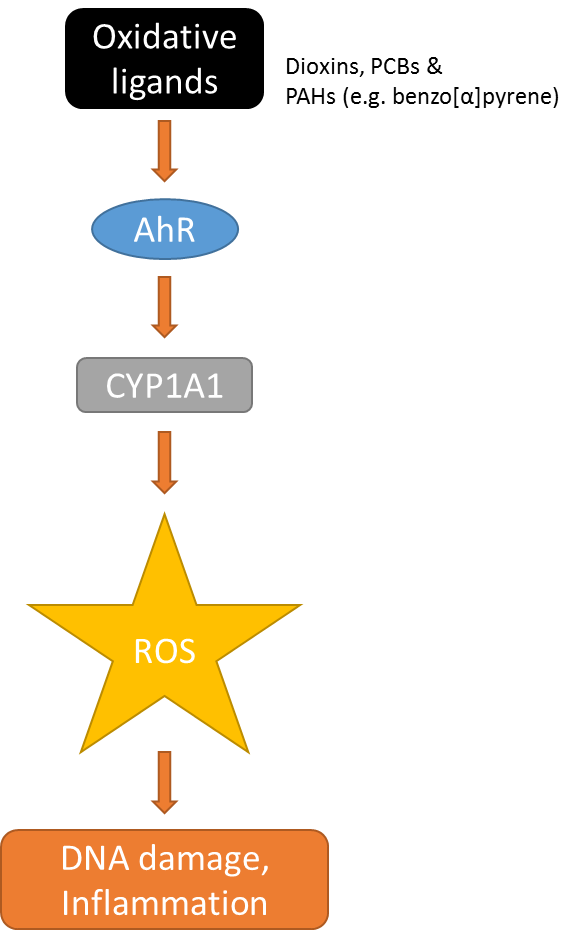
Las enzimas desintoxicantes genéticas, como el citocromo P-450A1, son capaces de hidrofilar sustratos hidrófobos. Mientras que esta reacción normalmente facilita la excreción de la sustancia modificada, algunos hidrocarburos aromáticos policíclicos (HAP), como el benzo [a] pireno, generan epóxidos semiestables que en última instancia pueden reaccionar con ADN formando aductos mutagénicos (ver Sección Metabolismo y defensa xenobióticos). El principal regulador del metabolismo de fase I en vertebrados, el receptor de hidrocarburos arílicos (AhR), es un jugador crucial en este proceso. Algunos HAP, dioxinas y algunos PCB (los llamados congéneres coplanares; ver sección Mezclas complejas) se unen y activan AhR y aumentan la actividad de las enzimas de fase I, incluido el citocromo P-450A1 (CYP1A1), en varias veces. Este aumento del metabolismo oxidativo potencia los efectos tóxicos de las sustancias que conducen a un aumento del daño e inflamación del ADN (Figura 7).
Efectos oxidativos sobre la regulación del crecimiento celular
La producción de ROS y el estrés oxidativo pueden actuar sobre la proliferación celular y la apoptosis. Se ha demostrado que los bajos niveles de ROS influyen en las vías de transducción de señales y alteran la expresión génica.
Figura en preparación
Figura 8. Papel de las ROS en la expresión génica alterada. Figura adaptada de Klaassen (2013).
Muchos xenobióticos, al aumentar los niveles celulares de oxidantes, alteran la expresión génica a través de la activación de vías de señalización incluyendo cascadas mediadas por AMPc, vías calcio-calmodulina, factores de transcripción como AP-1 y NF-κB, así como señalización a través de proteínas activadas por mitógenos (MAP) quinasas (Figura 8). La activación de estas cascadas de señalización finalmente conduce a una expresión génica alterada o a una serie de genes, incluidos los que afectan la proliferación, diferenciación y apoptosis.
Referencias
Boelsterli, U.A. (2002). Toxicología mecanicista: las bases moleculares de cómo los químicos interrumpen los objetivos biológicos. Prensa CRC.
Cuypers, A., Plusquin, M., Remans, T., Jozefczak, M., Keunen, E., Gielen, H.,..., Nawrot, T. (2010). Estrés por cadmio: un reto oxidativo. Biometales 23, 927-940.
Furue, M., Takahara, M., Nakahara, T., Uchi, H. (2014). Papel del sistema AHR/ARNt en la homeostasis cutánea. Archivos de Investigación Dermatológica 306, 769-779.
Klaassen, C.D. (2013). Toxicología de Casarett & Doull: La ciencia básica de los venenos, Octava Edición, McGraw-Hill Professional.
Niesink, R.J.M., De Vries, J. & Hollinger, M. A. (1996). Toxicología: Principios y Aplicaciones. Prensa CRC.
Thévenod, F. (2009). Cascadas de cadmio y señalización celular: ¿ser o no ser? Toxicología y Farmacología Aplicada 238, 221-239.
El herbicida paraquat induce estrés oxidativo debido a
- Interacción con la cadena de transporte de electrones.
Su implicación en el ciclo redox.
Interactúa con el glutatión.
Su implicación en la reacción de Fenton.
¿Qué biopolímeros pueden sufrir daños por especies reactivas de oxígeno?
solo ADN y proteínas
solo ADN y membranas
solo proteínas y membranas
ADN, proteínas y membranas
Dado: Los tres pasos de la peroxidación lipídica:
I iniciación
II propagación
III terminación
Pregunta: ¿En qué paso (s) está involucrado O 2 como reactivo o como producto?
Sólo yo
Sólo II
I y II
II y III
El ADN mitocondrial, en comparación con el ADN nuclear, es relativamente susceptible al daño oxidativo de la base.
¿Cuál de las alternativas dadas no es correcta?
El aumento de la susceptibilidad del ADN mitocondrial se debe a:
La proximidad del ADN mitocondrial a la cadena de transporte de electrones
El ADN mitocondrial no está protegido por histonas
Los niveles limitados de compuestos antioxidantes dentro de las mitocondrias
El sistema limitado de reparación de ADN mitocondrial
4.2.4. Citotoxicidad: compuestos xenobióticos que causan muerte celular
Autores: Frank Van Belleghem, Karen Smeets
Revisores: Timo Hamers, Bas J. Blaauboer
Objetivos de aprendizaje:
Deberías ser capaz de:
- nombrar los principales factores que causan la muerte celular,
- describir el proceso de necrosis y apoptosis,
- describir las diferencias morfológicas entre apoptosis y necrosis,
- explicar qué forma de muerte celular es causada por sustancias químicas.
Palabras clave: muerte celular, apoptosis, necrosis, activación de caspasa, transición de permeabilidad mitocondrial
Descripción
La citotoxicidad o toxicidad celular es el resultado de un daño macromolecular inducido por químicos (ver la sección sobre Inactivación de proteínas) o alteraciones mediadas por receptores (ver la sección Interacciones del receptor). Eventos iniciales como unión covalente a ADN o proteínas; pérdida de control de calcio o estrés oxidativo (ver las secciones sobre Estrés oxidativo I y II) pueden comprometer funciones celulares clave o desencadenar la muerte celular. La muerte celular es el último punto final de la lesión celular letal; y puede ser causada por compuestos químicos, células mediadoras (es decir, células asesinas naturales) o condiciones físicas/ambientales (es decir, radiación, presión, etc.). El proceso multietapa de muerte celular implica varios procesos regulados y puntos de control a pasar antes de que la célula finalmente llegue a un punto de no retorno, lo que lleva a la muerte celular programada o apoptosis, o a una forma más accidental de muerte celular, llamada necrosis. En esta sección se describe el propio proceso citotóxico, las pruebas de citotoxicidad in vitro se tratan en la sección de Pruebas de toxicidad humana - II. Pruebas in vitro.
Toxicidad química que conduce a la muerte celular
Las células pueden mantener activamente el ambiente intracelular dentro de un rango estrecho de parámetros fisiológicos a pesar de los cambios en las condiciones del entorno circundante. Este estado estacionario interno se llama homeostasis celular. La exposición a compuestos tóxicos puede comprometer la homeostasis y provocar lesiones. La lesión celular puede ser directa (primaria) cuando una sustancia tóxica interactúa con una o más moléculas diana de la célula (por ejemplo, daño a las enzimas de la cadena de transporte de electrones), o indirecta (secundaria) cuando una sustancia tóxica perturba el microambiente de la célula (por ejemplo, disminución del suministro de oxígeno o nutrientes). La lesión se denomina reversible cuando las células pueden someterse a reparación de adaptación para lograr un nuevo estado estable viable. Cuando la lesión persiste o se vuelve demasiado grave, se vuelve irreversible y la célula finalmente perece, terminando así funciones celulares como respiración, metabolismo, crecimiento y proliferación, dando como resultado la muerte celular (Niesink et al., 1996).
Los principales factores que determinan la ocurrencia de muerte celular son:
- la naturaleza y concentración del compuesto tóxico activo -en algunos casos un intermedio reactivo- y la disponibilidad de ese agente en el sitio de las moléculas diana;
- el papel de las moléculas diana en el funcionamiento de la célula y/o en el mantenimiento del microambiente;
- la efectividad de los mecanismos de defensa celular en la desintoxicación y eliminación de agentes activos, en la reparación del daño (primario) y en la capacidad de inducir proteínas que promuevan o inhiban el proceso de muerte celular.
Es importante darse cuenta de que también sustancias “inofensivas” como la glucosa o la sal pueden provocar lesiones celulares y muerte celular al alterar la homeostasis osmótica a concentraciones suficientes. Incluso una molécula esencial como el oxígeno causa lesión celular a presiones parciales suficientemente altas (ver las secciones sobre Estrés oxidativo I y II). Aparte de eso, todos los químicos ejercen “toxicidad basal” (también llamada “narcosis”) como se describe en el cuadro de texto “narcosis y daño de membrana” en la sección Toxicodinámica e Interacciones Moleculares.
Los principales tipos de muerte celular: necrosis y apoptosis
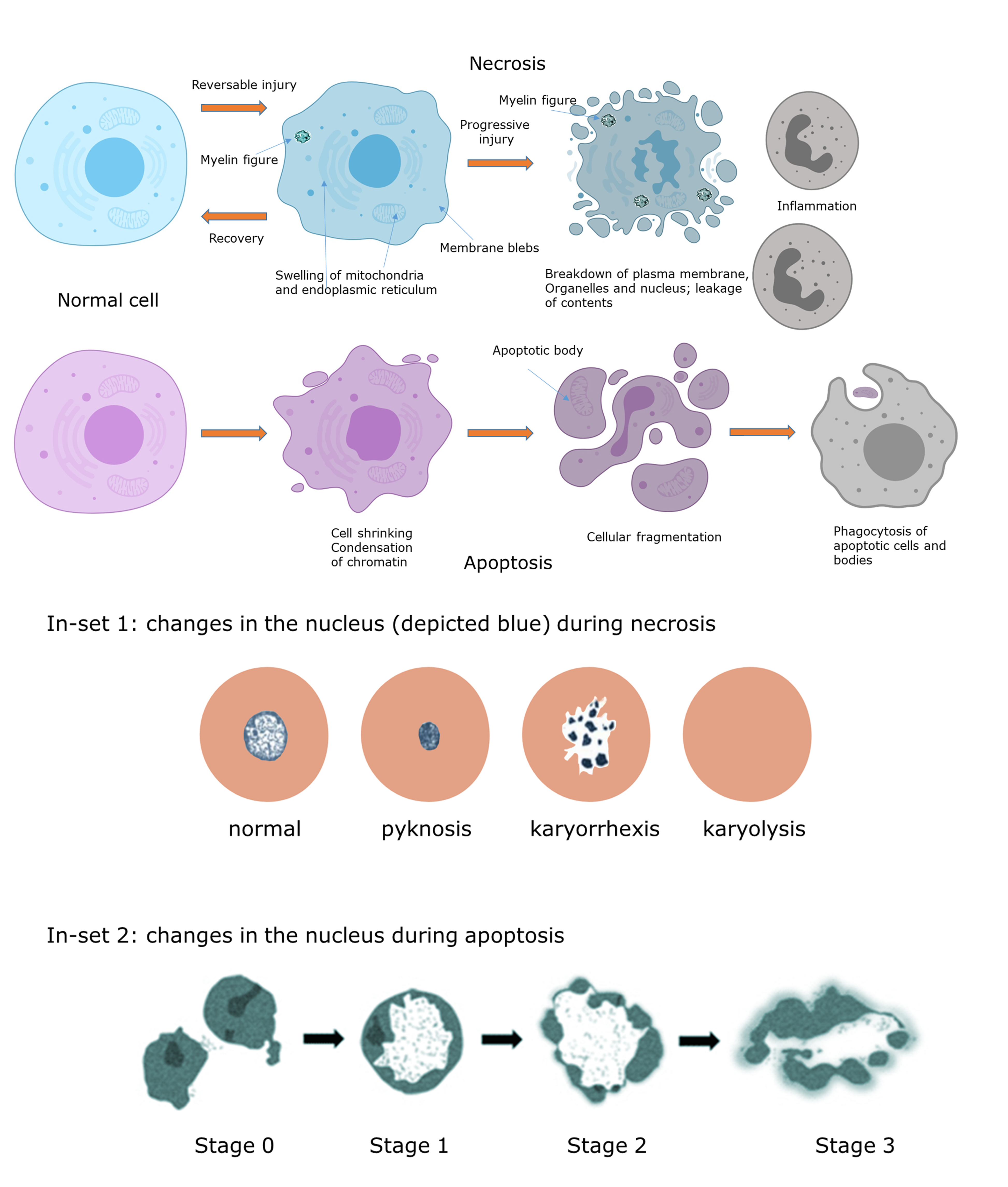
Los dos tipos más importantes de muerte celular son la necrosis o muerte celular accidental (ACD) y la apoptosis, una forma de muerte celular programada (PCD) o suicidio celular. Los desequilibrios celulares que inician o promueven la muerte celular solos o en combinación son estrés oxidativo, lesión mitocondrial o flujos alterados de calcio. Estas alteraciones son reversibles al principio, pero después de una lesión progresiva, resultan en la muerte celular irreversible. La muerte celular también puede iniciarse a través de procesos de transducción de señales mediados por receptores. Las células apoptóticas y necróticas difieren tanto en el aspecto morfológico como en las características bioquímicas. La necrosis se asocia con hinchazón celular y una rápida pérdida de integridad de la membrana. Las células apoptóticas se encogen en cuerpos apoptóticos Las células con fugas durante la necrosis inducen respuestas inflamatorias, aunque la inflamación no está completamente excluida durante el proceso apoptótico (Rock & Kono, 2008).
Necrosis La necrosis se ha denominado muerte celular accidental porque es una respuesta patológica a una lesión celular después de la exposición a estresores físicos, químicos o mecánicos severos. La necrosis es un proceso independiente de la energía que se corresponde con el daño a las membranas celulares y la posterior pérdida de homeostasis iónica (en particular Ca 2+). Esencialmente, la pérdida de integridad de la membrana celular permite que las enzimas se escapen de las membranas lisosómicas, destruyendo la célula desde el interior. La necrosis se caracteriza por hinchazón del citoplasma y orgánulos, ruptura de la membrana plasmática y condensación de cromatina (ver Figura 1). Estas apariencias morfológicas están asociadas con el agotamiento de ATP, defectos en la síntesis de proteínas, daño citoesquelético y daño al ADN. Además, los orgánulos celulares y los desechos celulares se filtran a través de las membranas dañadas al espacio extracelular, lo que lleva a la activación del sistema inmune y a la inflamación (Kumar et al., 2015). A diferencia de la apoptosis, la fragmentación del ADN es un evento tardío. En una etapa posterior, la lesión se propaga a través de los tejidos vecinos a través de la liberación de enzimas proteolíticas y lipolíticas dando como resultado áreas más grandes de tejido necrótico. Aunque la necrosis se considera tradicionalmente como una forma incontrolada de muerte celular, la evidencia emergente señala que el proceso también puede ocurrir de manera regulada y genéticamente controlada, denominada necrosis regulada (Berghe et al., 2014). Además, también puede ser un proceso autolítico de desintegración celular después de que se complete el programa apoptótico en ausencia de depuradores (fagocitos), denominados necrosis post-apoptótica o secundaria (Silva, 2010).
Apoptosis La apoptosis es un proceso fisiológico regulado (programado) mediante el cual se eliminan células superfluas o potencialmente dañinas (por ejemplo, células infectadas o precancerosas) de una manera estrechamente controlada. Es un proceso importante en el desarrollo embrionario, el sistema inmune y de hecho, todos los tejidos vivos. Las células apoptóticas se encogen y se rompen en pequeños fragmentos que son fagocitados por células adyacentes o macrófagos sin producir una respuesta inflamatoria (Figura 3). Se puede ver como una forma de suicidio celular porque la muerte celular es el resultado de la inducción de procesos activos dentro de la propia célula. La apoptosis es un proceso dependiente de la energía (requiere ATP) que implica la activación de caspasas (cisteína-aspartil proteasas), proteínas proapoptóticas presentes como zimógenos (es decir, precursores enzimáticos inactivos que se activan por hidrólisis). Una vez activadas, funcionan como cisteína proteasas y activan otras caspasas. Las caspasas se pueden distinguir en dos grupos, las caspasas iniciadoras, que inician el proceso, y las caspasas efectoras, que específicamente lisan moléculas que son esenciales para la supervivencia celular (Blanco & Blanco 2017). La apoptosis puede ser desencadenada por estímulos procedentes del interior de la célula (vía intrínseca) o del medio extracelular (vía extrínseca) como se muestra en la Figura 2. La vía extrínseca activa la apoptosis en respuesta a estímulos externos, es decir, por ligandos extracelulares que se unen a receptores de muerte de la superficie celular (Receptor del Factor de Necrosis Tumoral (TNFR)), lo que lleva a la formación del complejo de señalización inductor de muerte (DISC) y la cascada de caspasa que conduce a la apoptosis. La vía intrínseca es activada por factores estresantes celulares como daño en el ADN, falta de factores de crecimiento, estrés del retículo endoplásmico (ER), carga de especies reactivas de oxígeno (ROS), estrés de replicación, alteraciones microtubulares y defectos mitóticos (Galluzzi et al., 2018). Estos eventos celulares provocan la liberación del citocromo c y otras proteínas proapoptóticas de las mitocondrias al citosol a través del poro de transición de permeabilidad mitocondrial (MPT). Se trata de una megacanela en la membrana interna de las mitocondrias compuesta por varios complejos proteicos que facilitan la liberación de proteínas de muerte como el citocromo c. La apertura es desencadenada y fuertemente regulada por proteínas antiapoptóticas, como el linfoma-2 de células B (Bcl-2) y proteínas proapoptóticas, como Bax (proteína X asociada a Bcl-2) y Bak (antagonista asesino Bcl-2). Las vías intrínseca y extrínseca están reguladas por la proteína inhibidora de la apoptosis (AIP) que interactúa directamente con las caspasas y suprime la apoptosis. La liberación de la proteína de muerte citocromo c induce la formación de una gran estructura proteica formada en el proceso de apoptosis (el complejo apoptómico) activando la cascada de caspasa que conduce a la apoptosis. Otras proteínas proapoptóticas se oponen a Bcl (Smac/Diablo) y estimulan la actividad de la caspasa al interferir con AIP (hTRA2/Omi). Htra2/OMI también activa las caspasas y la endonucleasa G (responsable de la degradación del ADN, la condensación de la cromatina y la fragmentación del ADN). El factor inductor de apoptosis (AIF) está involucrado en la condensación de la cromatina y la fragmentación del ADN. Muchos xenobióticos interfieren con el poro MPT y el destino de una célula depende del equilibrio entre agentes pro y antiapoptóticos (Blanco & Blanco, 2017).
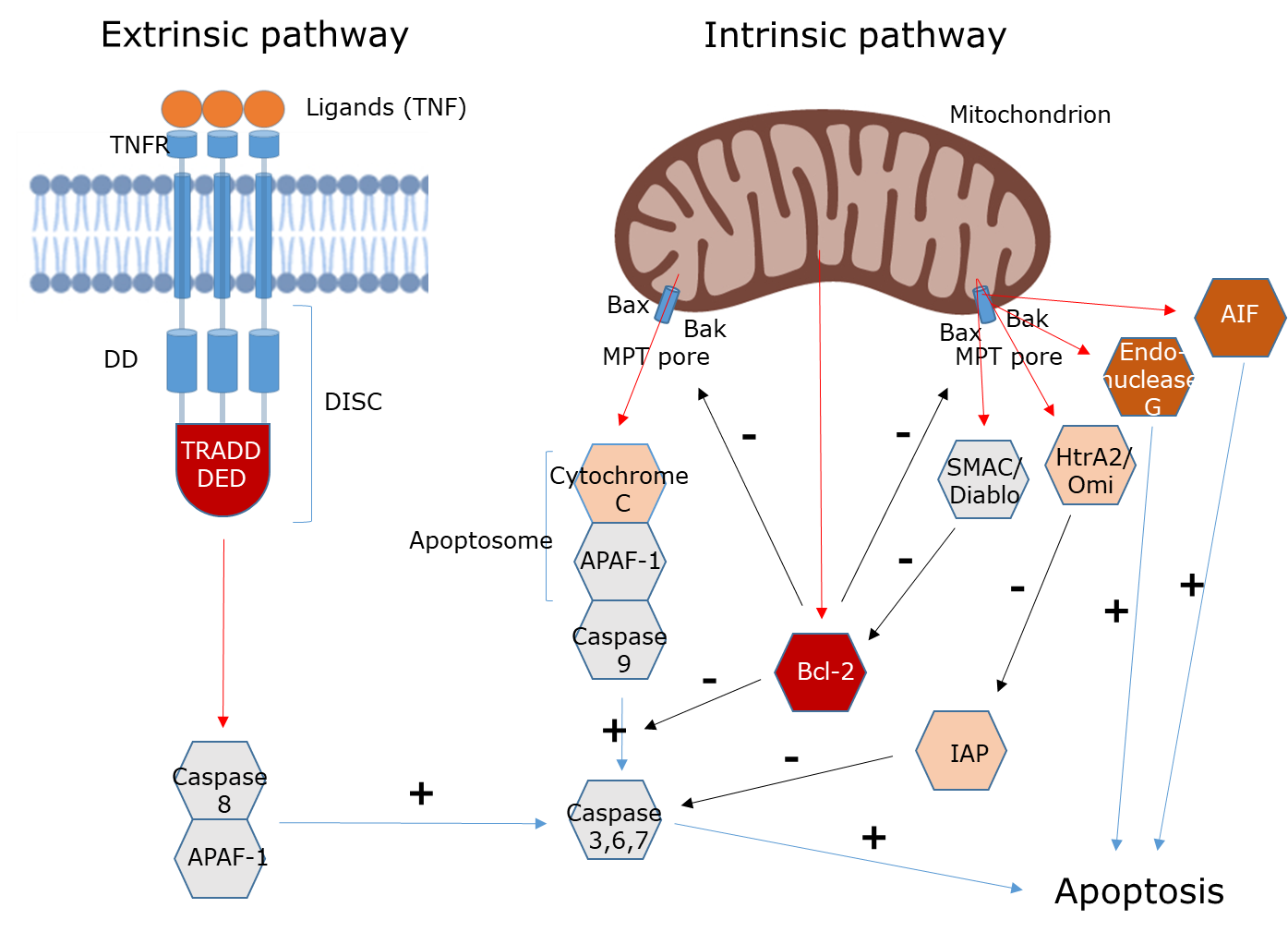
¿Qué determina la forma de muerte celular causada por sustancias químicas?
Tradicionalmente, se consideró que la muerte celular tóxica era única del tipo necrótico. El ejemplo clásico de necrosis es la toxicidad hepática del tetracloruro de carbono (CCl 4) causada por la biotransformación de CCl 4 a los radicales altamente reactivos (CCl 3 • y CCl 3 OO•).
Varios contaminantes ambientales, incluyendo metales pesados (Cd, Cu, CH 3 Hg, Pb), compuestos organoestánnicos y ditiocarbamatos, pueden ejercer su toxicidad a través de la inducción de la apoptosis, probablemente mediada por la alteración de la homeostasis intracelular de Ca 2+, o inducción de estrés oxidativo leve ( Orrenius et al., 2011).
Además, algunas sustancias citotóxicas (por ejemplo, trióxido de arsénico (As 2 O 3)) tienden a inducir apoptosis a niveles bajos de exposición o temprano después de la exposición a niveles altos, mientras que causan necrosis posteriormente a niveles altos de exposición. Esto implica que la gravedad del insulto determina el modo de muerte celular (Klaassen, 2013). En estos casos, tanto la apoptosis como la necrosis implican la disfunción de las mitocondrias, con un papel central para la transición de permeabilidad mitocondrial (MPT). Normalmente, la membrana mitocondrial es impermeable a todos los solutos excepto a los que tienen transportadores específicos. La MPT permite la entrada en las mitocondrias de solutos con un peso molecular inferior a 1500 Daltons, lo cual es causado por la apertura de poros de transición de permeabilidad mitocondrial (MPTP) en la membrana mitocondrial interna. A medida que estos solutos de masa molecular pequeña se equilibran a través de la membrana mitocondrial interna, el potencial de la membrana mitocondrial (δψMT) desaparece (despolarización mitocondrial), lo que lleva al desacoplamiento de la fosforilación oxidativa y posterior agotamiento del trifosfato de adenosina (ATP). Además, dado que las proteínas permanecen dentro de la matriz a alta concentración, la creciente presión osmótica coloidal dará como resultado el movimiento del agua hacia la matriz, lo que provoca hinchazón de las mitocondrias y ruptura de la membrana externa. Esto da como resultado la pérdida de componentes intermembranales (como citocromo c, AIF, Htra2/OMI, Smac/Diablo y Endonucleasa G) al citoplasma. Cuando la MPT ocurre en algunas mitocondrias, las mitocondrias afectadas se fagocitan y la célula sobrevive. Cuando se ven afectadas más mitocondrias, la liberación de compuestos pro-apoptóticos conducirá a la activación de la caspasa dando como resultado apoptosis. Cuando todas las mitocondrias están afectadas, el ATP se agota y la célula eventualmente sufrirá necrosis como se muestra en la Figura 3 (Klaassen et al., 2013).
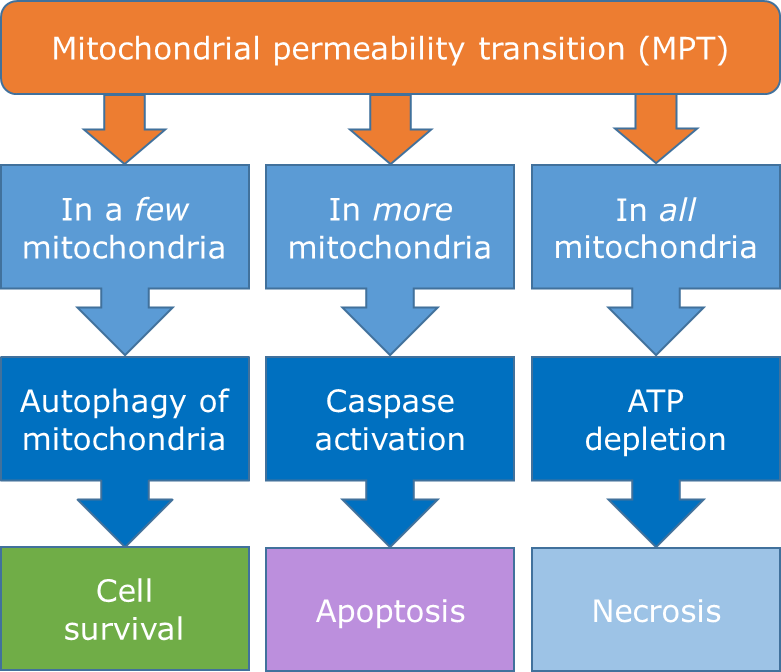
Referencias
Berghe, T.V., Linkermann, A., Jouan-Lanhouet, S., Walczak, H., Vandenabeele, P. (2014). Necrosis regulada: la red en expansión de vías de muerte celular no apoptóticas. Opiniones sobre la naturaleza Biología Celular Molecular 15, 135. https://doi.org/10.1038/nrm3737
Blanco, G., Blanco, A. (2017). Capítulo 32 - Apoptosis. Bioquímica médica. (pp. 791-796) Prensa Académica. https://doi.org/10.1016/B978-0-12-803550-4.00032-X
Galluzzi, L., Vitale, I., Aaronson, S.A., Abrams, J.M., Adam, D., Agostinis, P.,... & Annicchiarico-Petruzzelli, M. (2018). Mecanismos moleculares de la muerte celular: recomendaciones del Comité de Nomenclatura de Muerte Celular 2018. Muerte y diferenciación celular, 1. https://doi.org/10.1038/s41418-017-0012-4
Klaassen, C.D., Casarett, L.J., & Doull, J. (2013). Toxicología de Casarett y Doull: La ciencia básica de los venenos (8a ed.). Nueva York: McGraw-Hill Educación/Medicina.
ISBN: 978-0-07-176922-8
Kumar, V., Abbas, A.K., & Aster, J.C. (2015). Bases patológicas de la enfermedad de Robbins y Cotran, edición profesional. Elsevier Ciencias de la Salud. ISBN 978-0-323-26616-1.
Niesink, R.J.M., De Vries, J., Hollinger, M.A. (1996) Toxicología: principios y aplicaciones, (1ª ed.). Prensa CRC. ISBN 0-8493-9232-2.
Orrenius, S., Nicotera, P., Zhivotovsky, B. (2011). Mecanismos de muerte celular y sus implicaciones en toxicología. Ciencias Toxicológicas 119, 3-19. https://doi.org/10.1093/toxsci/kfq268
Rock, K.L., Kono, H. (2008). La respuesta inflamatoria a la muerte celular. Annú. Rev. Pathmechdis. Mech. Dis. 3, 99-126. https://doi.org/10.1146/annurev.pathmechdis.3.121806.151456
Silva, M.T. (2010). Necrosis secundaria: el resultado natural del programa apoptótico completo. FEBS Cartas 584, 4491-4499. doi.org/10.1016/j.febslet.2010.10.046
Toné, S., Sugimoto, K., Tanda, K., Suda, T., Uehira, K., Kanouchi, H.,... & Earnshaw, W. C. (2007). Tres etapas distintas de condensación nuclear apoptótica reveladas por imágenes de lapso de tiempo, análisis bioquímico y microscopía electrónica de la apoptosis libre de células. Investigación Celular Experimental 313, 3635-3644. https://doi.org/10.1016/j.yexcr.2007.06.018
¿Cuál de las siguientes es una característica de la necrosis?
- Los cambios morfológicos son causados por la liberación de enzimas lisosómicas.
- Es un proceso dependiente de la energía
- Es una respuesta programada a la lesión celular.
No conduce a la inflamación
¿Cuál de las siguientes es una característica de la apoptosis?
Formación de la membrana
Rápida pérdida de integridad de la membrana
Hinchazón de las mitocondrias
Encogimiento de celdas
¿Qué orgánulos celulares están involucrados en el inicio de la vía intrínseca de la apoptosis?
Ribosomas
Lisosomas
Mitocondrias
Peroxisomos
Considere las siguientes declaraciones:
- La necrosis secundaria es una forma de muerte celular accidental.
- La MPT permite la entrada de solutos conduciendo al incremento del volumen del citoplasma.
¿Qué afirmación es correcta?
- Sólo yo
- Sólo II
- Ni yo, ni II
- Tanto I como II
4.2.5. Neurotoxicidad
Autor: Jessica Legradi
Revisores: Timo Hamers, Ellen Fritsche
Objetivos de aprendizaje
Deberías ser capaz de
- describir la estructura del sistema nervioso
- explicar cómo funciona la neurotransmisión
- mencionar algunos modos de acción (MoA) por los cuales pesticidas y medicamentos causan neurotoxicidad
- entiende la relevancia de la sensibilidad de las especies a los plaguicidas
- describir qué es la neurotoxicidad del desarrollo (DNT)
Palabras clave: Sistema nervioso, Transmisión de señal, Plaguicidas, Medicamentos, Neurotoxicidad del desarrollo
Neurotoxicidad
La neurotoxicidad se define como la capacidad de los agentes para causar efectos adversos sobre el sistema nervioso. La neurotoxicidad ambiental describe la neurotoxicidad causada por la exposición a sustancias químicas del ambiente y se refiere principalmente a la exposición humana y la neurotoxicidad humana. La neurotoxicidad ecológica (econeurotoxicidad) se define como la neurotoxicidad resultante de la exposición a sustancias químicas ambientales en especies distintas de los humanos (por ejemplo, peces, aves, invertebrados).
El sistema nervioso
El sistema nervioso consiste en el sistema nervioso central (SNC) incluyendo el cerebro y la médula espinal y el sistema nervioso periférico (SNP). El SNP se divide en el sistema somático (movimientos voluntarios), el sistema autónomo (simpático y parasimpático) y el sistema entérico (gastrointestinal). El SNC y el SNP se construyen a partir de dos tipos de células nerviosas, es decir, neuronas y células gliales. Las neuronas son células que reciben, procesan y transmiten información a través de señales eléctricas y químicas. Las neuronas consisten en el soma con las dendritas circundantes y un axón con un axón terminal donde la señal se transmite a otra célula (Figura 1A). En comparación con las neuronas, las células gliales pueden tener apariencias muy diferentes (Figura 1B), pero siempre se encuentran en el tejido circundante de las neuronas donde proporcionan metabolitos, soporte y protección a las neuronas sin estar directamente involucradas en la transmisión de señales.
Figura en preparación
Figura 1. Estructuras de una neurona (izquierda; fuente: https://simple.Wikipedia.org/wiki/Neuron) y de células gliales (derecha)
Las neuronas están conectadas entre sí a través de sinapsis. La neurona emisora se llama neurona presináptica mientras que la neurona receptora es la neurona postsináptica. En la sinapsis, existe un pequeño espacio entre el axón terminal de la neurona presináptica y una dendrita de la neurona postsináptica. Este espacio se denomina hendidura sináptica. Ambas neuronas tienen canales iónicos que se pueden abrir y cerrar en la zona de la sinapsis. Hay canales selectivos para cloruro, sodio, calcio, potasio o protones y canales no selectivos. Los canales pueden ser controlados por voltaje (es decir, se abren y cierran dependiendo del potencial de la membrana), se pueden activar por ligando (es decir, se abren y cierran dependiendo de la presencia de otras moléculas que se unen al canal iónico), o pueden activarse por tensión (es decir, se abren y cierran debido al estrés físico (estiramiento)). Los ligandos que pueden abrir o cerrar canales iónicos se denominan neurotransmisores. Dependiendo del canal iónico y si se abre o cierra tras la unión del neurotransmisor, un neurotransmisor puede inhibir o estimular la despolarización de la membrana (es decir, neurotransmisor inhibidor o excitador, respectivamente). Los ligandos se unen al canal iónico a través de receptores (enlace a la sección sobre la interacción del receptor). Los neurotransmisores tienen funciones muy distintas y están vinculados a procesos físicos como la contracción muscular y el calor corporal y a procesos emocionales/cognitivos como ansiedad, placer, relajación y aprendizaje. La transmisión de la señal a través de la sinapsis (es decir, neurotransmisión) se ilustra en la Figura 2.
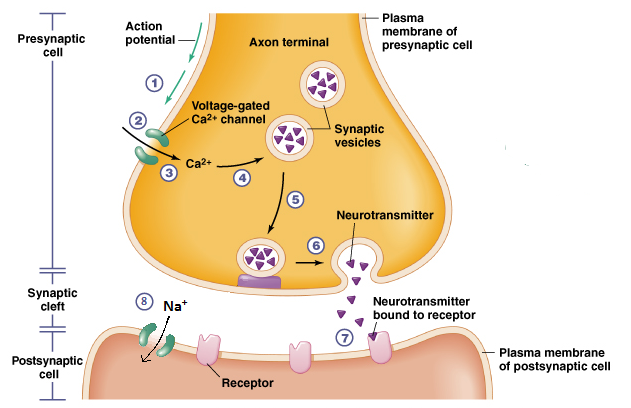
La membrana celular de una neurona contiene canales que permiten que los iones entren y salgan de la neurona. Este flujo de iones se utiliza para enviar señales de una neurona a otra. La diferencia en la concentración de iones cargados negativamente y positivamente en el lado interno y externo de la membrana neuronal crea un voltaje a través de la membrana llamado potencial de membrana. Cuando una neurona está en reposo (es decir, sin señalización), la carga interna de la neurona es negativa en relación con el exterior. La membrana celular se encuentra entonces en su potencial de reposo. Sin embargo, cuando una neurona está señalando, los cambios en el flujo de entrada y salida de iones conducen a una rápida despolarización seguida de una repolarización del potencial de membrana llamado potencial de acción. Aquí se puede encontrar un video que muestra cómo se produce el potencial de acción.
Las neuronas pueden dañarse a través de sustancias que dañan el cuerpo celular (neuronopatía), el axón (axonopatía), o la lámina de mielina o las células gliales (mielopatía). El aluminio, el arsénico, el metanol, el metilmercurio y el plomo pueden causar neuropatía. Se sabe que la acrilamida afecta específicamente a los axones y causa axonopatía.
Modos de acción de neurotoxicidad relacionados con el sistema neurotransmisor
Algunos de los modos de acción relevantes para la neurotoxicidad son las alteraciones de la transmisión de señales eléctricas y la inhibición de la transmisión de señales químicas, principalmente a través de la interferencia con los neurotransmisores. Los pesticidas están diseñados principalmente para interferir con la neurotransmisión.
1. Interferir con los canales iónicos (ver sección sobre Interacción del receptor )
Los pesticidas como el DDT se unen a canales abiertos de sodio en las neuronas, lo que impide el cierre de los canales y conduce a la sobreexcitación. Los piretroides, como la permetrina, aumentan el tiempo de apertura de los canales de sodio, lo que lleva a síntomas similares. Lindano, insecticidas ciclodiénicos como aldrina, dieldrina y endrina (“drins”) y fenil-pirazoles como el fipronil bloquean los canales de cloruro mediados por GABA y previenen la hiperpolarización. El GABA (ácido gamma-aminobutírico) es un neurotransmisor inhibidor que está vinculado a la relajación y la calma. Estimula la apertura de canales de cloruro haciendo que el potencial transmembrana se vuelva más negativo (es decir, hiperpolarización), aumentando así el umbral de despolarización para un nuevo potencial de acción. Los bloqueadores de los canales de cloruro mediados por GABA previenen el efecto hiperpolarizante del GABA, disminuyendo así el efecto inhibitorio del GABA. Los neonicotinoides (e.g., imidacloprid) imitan la acción del neurotransmisor excitatorio ACh activando los receptores nicotínicos de acetilcolina (nAChR) en la membrana postsináptica. Estos compuestos están diseñados específicamente para mostrar una alta afinidad por nAChR de insectos.
Muchos fármacos humanos, como los sedantes, también se unen a los neuroreceptores. Los fármacos benzodiazepinos activan los receptores GABA causando hiperpolarización (activando GABA). El tetrahidrocannabinol (THC), que es el ingrediente activo del cannabis, activa los receptores cannabinoides provocando también hiperpolarización. Los compuestos que activan los receptores GABA o cannabinoides inducen una fuerte sensación de relajación. La nicotina se une y activa el AChR, lo que puede ayudar a concentrarse.
2. Inhibición AChE
Otro modo de acción neurotóxico muy común es la inhibición de la acetilcolinesterasa (AChE). Insecticidas organofosforados como diclorvos e insecticidas carbamato como propoxur se unen a la AChE, y por lo tanto previenen la degradación de la acetilcolina en la hendidura sináptica, lo que lleva a la sobreexcitación de la membrana celular possináptica (ver también sección sobre Interacción de proteínas).
3. Bloqueo de la absorción de neurotransmisores
MDMA (3,4-metilendioximetanfetamina, también conocida como éxtasis o XTC) y cocaína bloquean la recaptación de serotonina, norepinefrina y en menor cantidad dopamina en la neurona presináptica, aumentando así la cantidad de estos neurotransmisores en la hendidura sináptica. Las anfetaminas también aumentan la cantidad de dopamina en la hendidura al estimular la liberación de dopamina de las vesículas. La dopamina es un neurotransmisor que está involucrado en el placer y en los sentimientos de recompensa. La serotonina o 5-hidroxitriptamina es un neurotransmisor monoamínico ligado a sentimientos de felicidad, aprendizaje, recompensa y memoria.
Exposición a largo plazo
Cuando los receptores se activan continuamente o cuando los niveles de neurotransmisores se elevan continuamente, el sistema nervioso se adapta volviéndose menos sensible al estímulo. Esto explica por qué los drogadictos tienen que aumentar el número de drogas que se toman para llegar al estado deseado. Si no se toma ningún estimulante, los síntomas de abstinencia ocurren por la falta de estímulo. En la mayoría de los casos, el sistema nervioso puede recuperarse de la drogadicción.
Sensibilidad de Especies en Neurotoxicidad
Las diferencias en la sensibilidad de las especies pueden explicarse por diferencias en las capacidades metabólicas entre especies. La mayoría de los compuestos necesitan ser bioactivados, es decir, ser biotransformados en un metabolito que cause el efecto tóxico real. Por ejemplo, la mayoría de los insecticidas organofosforados son tio-fosfoésteres que requieren oxidación antes de provocar la inhibición de la AChE. Como la desintoxicación es la vía dominante en los mamíferos y la oxidación es la vía dominante en los invertebrados, los insecticidas organofosforados suelen ser más tóxicos para los invertebrados que para los vertebrados (ver Figura 3). Otros factores importantes para la sensibilidad de las especies son la absorción y la tasa de depuración.
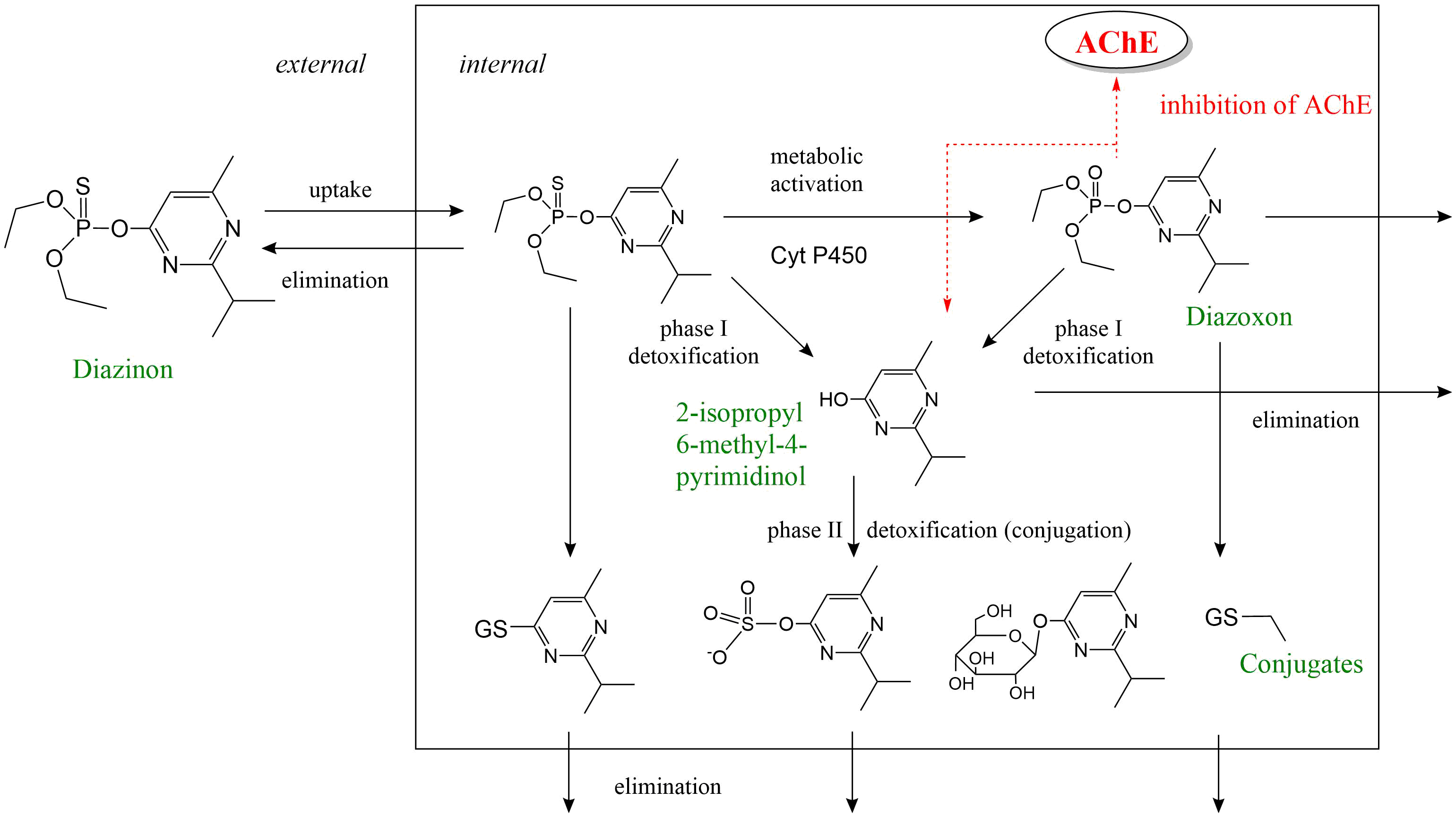
Neurotoxicidad del desarrollo
La neurotoxicidad del desarrollo (DNT) se refiere particularmente a los efectos de los tóxicos sobre el desarrollo del sistema nervioso de los organismos. Se supone que el cerebro y el sistema nervioso en desarrollo son más sensibles a los efectos tóxicos que el cerebro y el sistema nervioso maduros. Los estudios DNT deben considerar la ocurrencia temporal y regional de procesos críticos de desarrollo del sistema nervioso, y el hecho de que la exposición temprana de la vida puede conducir a efectos neurotóxicos duraderos o retrasos en el desarrollo neurológico. Las diferencias de especies también son relevantes para DNT. Aquí, el tiempo de desarrollo, la velocidad o las especificidades celulares podrían determinar la toxicidad.
¿Cuáles son los dos principales tipos de células que se encuentran en el sistema nervioso?
¿Qué hace GABA?
¿Cómo funciona la inhibición de AChE?
¿Qué hace que los invertebrados sean más sensibles a los insecticidas organofosforados?
¿Por qué es importante estudiar DNT?
4.2.6. Efectos de los herbicidas
Autor: Nico M. van Straalen
Revisores: Cornelia Kienle, Henk Schat
Objetivos de aprendizaje
Deberías ser capaz de
- Explicar las diferentes formas en que se aplican los herbicidas en la agricultura moderna
- Enumerar los ocho principales modos de acción de los herbicidas
- Proporcionar algunos ejemplos de efectos secundarios de los herbicidas
Palabras clave: Inhibidor de aminoácidos, regulador del crecimiento, inhibidor de la fotosíntesis, aplicación pre-emergencia, selectividad
Introducción
Los herbicidas son pesticidas (ver sección Productos de protección de cultivos) que tienen como objetivo matar malezas no deseadas en sistemas agrícolas, y malas hierbas que crecen en infraestructura como pavimento y vías de tren. Los herbicidas también se aplican al propio cultivo, por ejemplo, como tratamiento previo a la cosecha en cultivos como papa y colza oleaginosa, para prevenir el crecimiento de patógenos en plantas más viejas, o para facilitar la cosecha mecánica. De manera similar, los herbicidas se utilizan para destruir pasto de pastos en preparación de su conversión a tierras de cultivo. Estas aplicaciones se denominan “desecación”. Finalmente, se utilizan herbicidas para matar malezas de hoja ancha en campos de pasto puro (por ejemplo, canchas de golf).
Los herbicidas representan el mayor volumen de pesticidas aplicados hasta la fecha (aproximadamente 60%), en parte porque el control mecánico y manual de malezas ha disminuido considerablemente. La tendencia a limitar la labranza del suelo (como estrategia para mantener una vida diversa y saludable del suelo) también ha estimulado el uso de herbicidas químicos.
Los herbicidas obviamente están diseñados para matar plantas y, por lo tanto, actuar sobre dianas bioquímicas que son específicas de las plantas. Como el cultivo en sí también es una planta, la selectividad es un tema muy importante en la aplicación de herbicidas. Esto se logra de varias maneras.
- Aplicación de herbicidas antes de la emergencia del cultivo (aplicación pre-emergencia). Esto mantendrá el campo libre de malezas antes de la germinación, mientras que el dosel cerrado del cultivo impide el crecimiento posterior de malezas. Esta estrategia suele aplicarse en cultivos de rápido crecimiento que hacen un dosel alto con mucha sombra a nivel del suelo, como el maíz. Ejemplos de herbicidas utilizados en la aplicación pre-emergencia son glifosato y metolaclor. También la selectividad de los inhibidores del crecimiento de plántulas como el EPTC se debe a que estos compuestos se aplican como parte de una preparación del suelo y actúan sobre la germinación de las plantas antes de que emerja el cultivo.
- Las plantas de hoja ancha son más susceptibles a los herbicidas que dependen del contacto con las hojas, porque interceptan más un aerosol herbicida que las plantas de hoja pequeña como el pasto. Este tipo de selectividad permite que algunos herbicidas sean utilizados en cultivos de pastizales y cereales, para controlar malezas de hoja ancha; el herbicida en sí no es interceptado por el cultivo. Ejemplos son los ácidos clorofenoxi-acéticos tales como MCPA y 2,4-D.
- En algunos casos la planta de cultivo es naturalmente tolerante a un herbicida debido a vías metabólicas específicas. La selectividad de los inhibidores de ACCasa como diclofop-metilo, fenoxaprop-etilo y fluazifop-butilo se debe principalmente a este mecanismo. Estos compuestos inhiben las acetil-CoA carboxilasas, un grupo de enzimas esenciales para la síntesis de ácidos grasos. Sin embargo, en el trigo los compuestos herbicidas se hidrolizan rápidamente a metabolitos no tóxicos, mientras que las malas hierbas no son capaces de tal desintoxicación. Esto permite que dichos herbicidas sean utilizados en campos de trigo. Otro tipo de selectividad fisiológica se debe a la translocación diferencial, es decir, algunas plantas transportan rápidamente el herbicida por toda la planta, lo que le permite ejercer toxicidad en las hojas, mientras que otras mantienen la sustancia en las raíces y así permanecen menos susceptibles.
- Varios cultivos han sido modificados genéticamente (gm) para volverse resistentes a herbicidas; una de las modificaciones más conocidas es la inserción de una versión alterada de la enzima EPSP sintasa. Esta enzima forma parte de la vía del shikimato y es inhibida específicamente por el glifosato (Figura 1). La versión modificada de la enzima hace que la planta sea insensible al glifosato, permitiendo el uso de herbicidas sin dañar el cultivo. Varias especies de plantas han sido modificadas de esta manera, aunque su cultivo se limita a países que permiten cultivos transgénicos (Estados Unidos y muchos otros países, pero no países europeos).
Clasificación por modo de acción
La diversidad de compuestos químicos que se han sintetizado para atacar dianas bioquímicas específicas en las plantas es enorme. En un intento de clasificar herbicidas por modo de acción, a menudo se utiliza un sistema de 22 categorías diferentes (Sherwani et al. 2015). Aquí presentamos una clasificación simplificada que especifica solo ocho categorías (Plant & Soil Sciences eBrary 2019, Tabla 1).
Cuadro 1. Clasificación de herbicidas por modo de acción
|
No. |
Clase (modo de acción) |
Ejemplos de grupos químicos |
Ejemplo de ingrediente activo |
|
1 |
Inhibidores de síntesis de aminoácidos |
Sulfonilureas, imidazolonas, triazolopirimidinas, inhibidores de epsp sintasa |
Glifosato |
|
2 |
Inhibidores del crecimiento de plántulas |
Carbamotiatos, acetamidas, dinitroanilinas |
EPTC |
|
3 |
Reguladores del crecimiento (interfieren con las hormonas vegetales) |
Ácidos fenoxi-acéticos, ácido benzoico, ácidos carboxílicos, ácidos picolínicos |
2,4-D |
|
4 |
Inhibidores de la fotosíntesis |
Triazinas, uracilos, fenilureas, benzotiadiazoles, nitrilos, piridazinas |
Atrazina |
|
5 |
inhibidores de la síntesis lipídica |
Ariloxifenoxipropionatos, ciclohexanodionas |
Sethoxidim |
|
6 |
Disruptores de membrana celular |
Difenileteres, aril triazolinonas, fenilftalamidas, bipiridilio |
Paraquat |
|
7 |
Inhibidores de pigmentos protectores |
Isoxazolidonas, isoxazoles, piridazinonas |
Mesotriona |
|
8 |
Desconocido |
Compuestos químicos con eficacia herbicida comprobada pero modo de acción desconocido |
Etofumesato |
Para ilustrar la diversidad de modos de acción herbicidas, se destacan aquí dos ejemplos de mecanismos bien investigados.
Las plantas sintetizan aminoácidos aromáticos usando la vía del shikimato. También bacterias y hongos hacen uso de esta vía, pero no está presente en animales. Deben obtener aminoácidos aromáticos a través de su dieta. El primer paso en esta vía es la conversión de shikimato-3-fosfato y fosfoenolpiruvato (PEP) a 5-enolpiruvilshikimato-3-fosfato (EPSP), por la enzima EPSP sintasa (Figura 1). EPSP se desfosforila posteriormente y forma el sustrato para la síntesis de aminoácidos aromáticos como fenilalanina, tirosina y triptófano.
El glifosato tiene un parecido estructural con PEP y compite con PEP como sustrato para la EPSP sintasa. Sin embargo, a diferencia de PEP, se une firmemente al sitio activo de la enzima y bloquea su actividad. La consiguiente deficiencia metabólica conduce rápidamente a la pérdida del potencial de crecimiento de la planta.
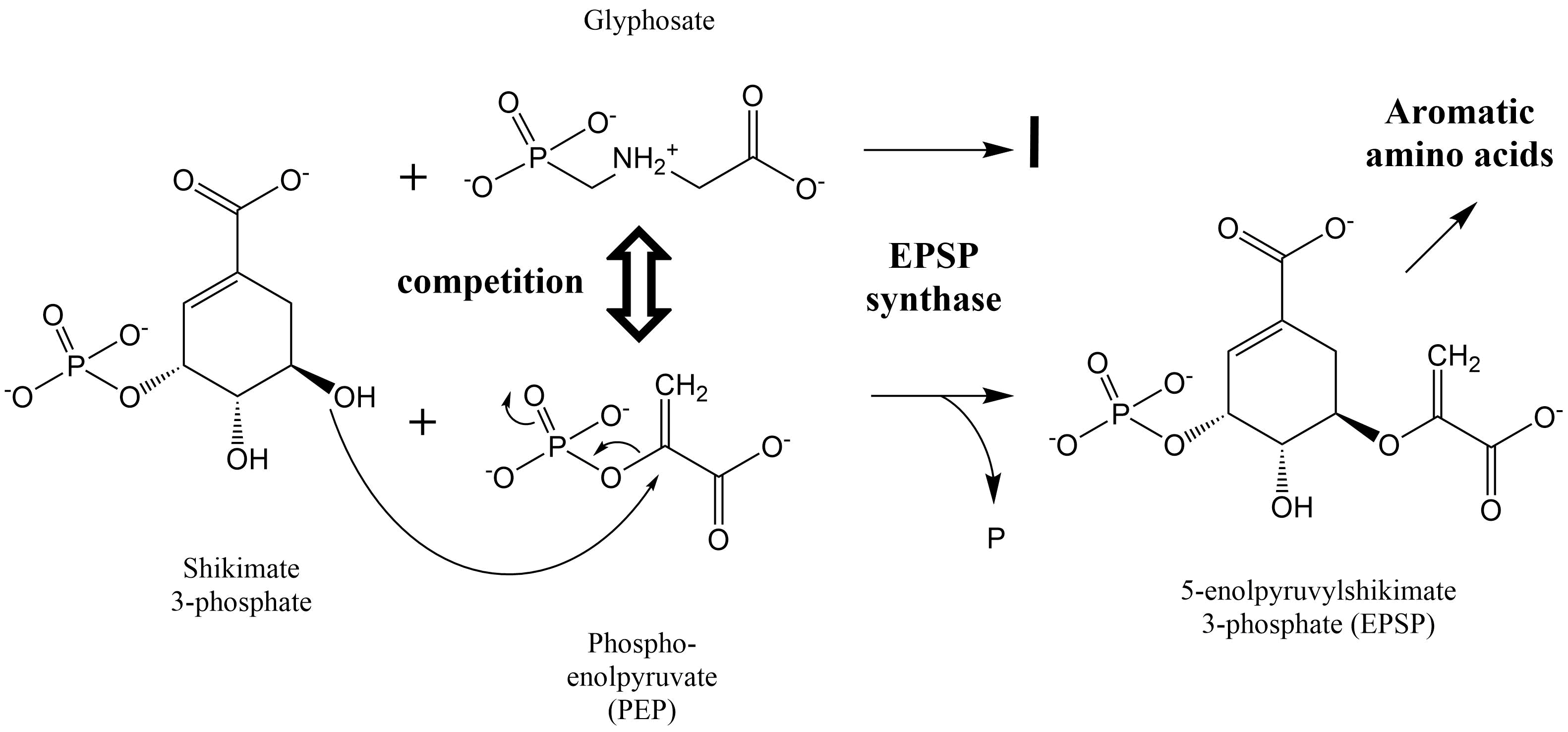
Otro modo de acción herbicida muy bien investigado es la inhibición de la fotosíntesis por atrazina y otras triazinas simétricas. A diferencia del glifosato, la atrazina solo puede actuar en plantas aéreas con fotosíntesis activa. El clima soleado estimula la acción de tales herbicidas. La acción de la atrazina se debe a la unión a la proteína quinona D1 del complejo de transporte de electrones del fotosistema II que se encuentra en la membrana interna del cloroplasto (ver Figura 1 en Giardi y Pace, 2005). El fotosistema II (PSII) es un complejo de macromoléculas con unidades de captación de luz y antena, clorofila P680 y centros de reacción que capturan la energía de la luz y la utilizan para dividir el agua, producir oxígeno y transferir electrones al fotosistema I, que los utiliza para producir eventualmente equivalentes de reducción. La quinona D1 tiene un “bolsillo de unión a herbicida” y la unión de la atrazina a este sitio bloquea la función del PSII. Un solo aminoácido en el bolsillo de unión es crítico para esto; las alteraciones en este aminoácido proporcionan una posibilidad relativamente fácil de que la planta se vuelva resistente a las triazinas.
Efectos secundarios
La mayoría de los herbicidas son compuestos polares con buena solubilidad en agua, lo que es una propiedad crucial para que sean captados por las plantas. Esto implica que los herbicidas, especialmente los más persistentes, tienden a lixiviarse a las aguas subterráneas y superficiales y a veces también se encuentran en los recursos de agua potable. Dados los grandes volúmenes aplicados en la agricultura, ha surgido la preocupación de que tales compuestos, a pesar de que están diseñados para afectar únicamente a las plantas, puedan dañar a otros organismos llamados “no diana”.
En los sistemas agrícolas y sus alrededores inmediatos, la eliminación completa de malezas reducirá la biodiversidad vegetal, con efectos secundarios en insectos que se alimentan de plantas y aves insectívoras. A corto plazo sin embargo, los herbicidas aumentarán la cantidad de restos de plantas muertas en el suelo, lo que puede beneficiar a los invertebrados que son menos susceptibles al efecto herbicida, y encontrar refugio en la hojarasca de las plantas y alimentarse de materia orgánica muerta. Los estudios muestran que a menudo hay un efecto positivo de los herbicidas sobre Colémbola, ácaros y otros artrópodos tensoactivos (por ejemplo, Fratello et al. 1985). Otros efectos secundarios pueden ocurrir cuando los herbicidas alcanzan zanjas limítrofes de campo, donde la supresión de macrófitos y algas puede afectar a poblaciones de macroinvertebrados como gammaridos y caracoles.
Se espera toxicidad directa a organismos no diana de herbicidas de amplio espectro que matan plantas debido a un mecanismo general de toxicidad. Esto es válido para el paraquat, un herbicida de bipiridilio (ver Tabla 1) que actúa como agente de contacto y daña rápidamente las hojas de las plantas mediante el ciclo rédox; potenciado por la luz solar, genera radicales de oxígeno que interrumpen las membranas biológicas. El paraquat es obviamente tóxico para toda la vida y representa un peligro agudo para los humanos. En consecuencia, su uso como herbicida está prohibido en la UE desde 2007.
En otros casos la situación es más compleja. El glifosato, el herbicida con mucho el mayor volumen de aplicación a nivel mundial, es sospechoso de efectos secundarios ecológicos e incluso ha sido etiquetado como “un probable carcinógeno” por la UICR (Tarazona et al., 2017). Sin embargo, el glifosato es un ingrediente activo contenido en diversas formulaciones herbicidas, por ejemplo, Roundup Ready, Roundup 360 plus, etc. La evidencia indica que la mayor parte de la toxicidad atribuida al glifosato se debe en realidad a adyuvantes en la formulación, específicamente seboaminas polietoxiladas (Mesnage et al., 2013 ).
Otro caso de un efecto secundario inesperado de un herbicida es debido a la atrazina. En 2002 un grupo de ecologistas estadounidenses (Hayes et al., 2002) reportaron que la incidencia de anomalías del desarrollo en ranas silvestres se correlacionó con el volumen de atrazina que se vende en la zona donde se monitorearon las ranas, a través de una gran cantidad de sitios en los pipiens Rana macho de Estados Unidos expuestos a La atrazina en concentraciones superiores a 0.1 µg/L durante sus estadios larvales mostró una mayor tasa de feminización, es decir, el desarrollo de ovocitos en los testículos. Esto sería debido a la inducción de aromatasa, una actividad del citocromo P450 responsable de la conversión de la testosterona en estradiol.
Finalmente, el desarrollo de resistencia también puede considerarse un efecto secundario indeseable. Actualmente hay (2018) 499 casos únicos (255 especies de plantas, combinadas con 167 ingredientes activos) de resistencia a herbicidas, lo que indica la gravedad agronómica de este tema. Una discusión completa de este tema cae, sin embargo, más allá del alcance de este módulo.
Conclusiones
Los herbicidas son actualmente un componente indispensable y de alto volumen de la agricultura moderna. Representan un gran número de grupos químicos y diferentes modos de acción, a menudo específicos de plantas. Si bien algunos de los herbicidas más antiguos (paraquat, atrazina, glifosato) han suscitado preocupación por sus efectos adversos en dianas no vegetales, el desarrollo de nuevos químicos y el descubrimiento de nuevas dianas bioquímicas en vías metabólicas específicas de plantas sigue siendo un campo activo de investigación.
Referencias
Fratello, B. et al. (1985) Efectos de la atrazina en microartrópodos de suelo en campos experimentales de maíz. Pedobiologia 28:161-168.
Giardi, M.T., Pace, E. (2005) Proteínas fotosintéticas para aplicaciones tecnológicas. Tendencias en Biotecnología 23, 257-263.
Hayes, T., Haston, K., Tsui, M., Hoang, A., Haeffele, C., Vonk, A. (2002). Feminización de ranas macho en estado salvaje. Naturaleza 419, 895-896.
Mesnage, R., Bernay, B., Séralini, G.-E. (2013). Los adyuvantes etoxilados de herbicidas a base de glifosato son principios activos de toxicidad celular humana. Toxicología 313, 122-128.
Biblioteca electrónica de ciencias de plantas y suelos (2019), https://passel.unl.edu.
Sherwani, S.I., Arif, I.A., Khan, H.A. (2015). Modos de acción de diferentes clases de herbicidas. En: Price, J., Kelton, E., Suranaite, L. (Eds.). Herbicidas. Acción Fisiológica y Seguridad. Capítulo 8, INTECHOPEN.
Tarazona, J.V., Corte-Marques, D., Tiramani, M., Reich, H., Pfeil, R., Istace, F., Crivellente, F. (2017). Toxicidad y carcinogenicidad del glifosato: una revisión de las bases científicas de la Evaluación de la Unión Europea y sus diferencias con la IARC. Archivos de Toxicología 91, 2723-2743.
Definir qué se entiende por herbicida preemergencia y ¿por qué esto es útil en agronomía?
Con una aplicación herbicida en agricultura se quiere matar plantas no deseadas entre un cultivo que en sí mismo también es una planta. ¿Cómo es esto posible?
Enumerar los ocho principales modos de acción de los herbicidas
¿Pueden los herbicidas causar efectos adversos en especies no vegetales?
4.2.7. Carcinogénesis química y genotoxicidad
Autor: Timo Hamers
Crítico: Frederik-Jan van Schooten
Objetivos de aprendizaje
Deberías ser capaz de
- describir las tres fases diferentes en el desarrollo del cáncer y entender cómo los compuestos pueden estimular los procesos correspondientes en estas fases
- explicar la diferencia entre las sustituciones de pares de bases y las mutaciones de desplazamiento de marco tanto a nivel de ADN como de proteína
- describir el principio de bioactivación, que distingue las sustancias mutágenas indirectas de las directas
- explicar la diferencia entre carcinógenos mutagénicos y no mutagénicos
Palabras clave: Bioactivación; Mutación; Promoción tumoral; Progresión tumoral; Prueba de Ames
Carcinogénesis química
El cáncer es un nombre colectivo para múltiples enfermedades que comparten un fenómeno común de que la división celular está fuera de control por procesos reguladores del crecimiento. Las consiguientes células de crecimiento autónomo suelen concentrarse en una neoplasia (a menudo referida a un tumor) pero también pueden dispersarse difusamente, por ejemplo en caso de leucemia o mesotelioma. Los tumores benignos se refieren a neoplasias que están encapsuladas y no se distribuyen a través del cuerpo, mientras que los tumores malignos causan metástasis, es decir, diseminación de células cancerígenas a través del cuerpo provocando nuevas neoplasias a distancia. El término benigno suena más amigable de lo que realmente es: los tumores benignos pueden ser muy dañinos para órganos que están limitados en el espacio disponible (por ejemplo, el cerebro en el cráneo) o para órganos que pueden ser obstruidos por el tumor (por ejemplo, el sistema intestinal).
El proceso de desarrollo del cáncer (carcinogénesis) se divide tradicionalmente en tres fases, i.e.
- la fase de iniciación, en el ADN genético de una célula se cambia permanentemente, dando como resultado células hijas que difieren genéticamente de sus células parentales;
- la fase de promoción, en la que la célula pierde su diferenciación y adquiere nuevas características provocando una mayor proliferación;
- la fase de progresión, en la que el tumor invade los tejidos circundantes y provoca metástasis.
La carcinogénesis química significa que una sustancia química es capaz de estimular una o más de estas fases. Los compuestos cancerígenos a menudo reciben el nombre de la fase en la que afectan, es decir, iniciadores (también llamados mutágenos), promotores tumorales y progresores tumorales. Es importante darse cuenta de que muchas sustancias y procesos que ocurren naturalmente en el cuerpo también pueden estimular las diferentes fases, es decir, la inflamación y la exposición a la luz solar pueden causar mutaciones, algunas hormonas endógenas pueden actuar como promotoras muy activas en cánceres sensibles a hormonas, y mutaciones espontáneas puede estimular la fase de progresión tumoral.
Mutaciones puntuales
Las mutaciones génicas (también conocidas como mutaciones puntuales) son cambios permanentes en el orden de los pares de bases de nucleótidos en el ADN. Con base en lo que sucede a nivel del ADN, las mutaciones puntuales se pueden dividir en tres tipos, es decir, un reemplazo de un par de bases original por otro par de bases (sustitución de pares de bases), la inserción de un par de bases extra o la deleción de un par de bases original (Figura 1). En una parte codificante del ADN, tres nucleótidos adyacentes en una cadena de ADN (es decir, un triplete) forman un codón que codifica un aminoácido en la proteína final. Debido a que las inserciones y deleciones provocan un cambio en estos marcos de lectura de tripletes con un nucleótido a la izquierda o a la derecha, respectivamente, estas mutaciones puntuales también se denominan mutaciones de desplazamiento de marco.
Con base en lo que sucede a nivel proteico para el que codifica un gen, las mutaciones puntuales también se pueden dividir en tres tipos. Una mutación sin sentido significa que el gen mutado codifica una proteína diferente al gen de tipo silvestre, una mutación sin sentido significa que la mutación introduce un codón STOP que interrumpe la transcripción génica dando como resultado una proteína truncada, y una mutación silenciosa significa que el gen mutado todavía codifica exactamente la misma proteína, a pesar de que se ha cambiado el código genético. Las mutaciones silenciosas son siempre sustituciones de pares de bases, debido a que la estructura triplete del ADN no ha sido dañada.
|
Un ejemplo muy ilustrativo de la diferencia entre una sustitución de pares de bases y una mutación de desplazamiento de marco a nivel de expresión proteica es la siguiente oración “de tipo silvestre”, que consta de solo tres palabras de letras que representan los tripletes en el ADN genómico: El gato gordo se comió el hot dog. Imagínese que la letra t en cat es reemplazada por una r debido a una sustitución de pares de bases. La frase dice entonces: El auto gordo se comió el hot dog. Esta frase claramente tiene otro significado, es decir, contiene información de sentido erróneo. Imagínese ahora que la letra a en grasa es reemplazada por una e debido a una sustitución de pares de bases. La frase dice entonces: El gato fet se comió el hot dog. Esta frase contiene claramente un error ortográfico (es decir, una mutación), pero su significado no ha cambiado, es decir, contiene una mutación silenciosa. Imagínese ahora que una letra m adicional provoca un desplazamiento de marco en la palabra grasa, debido a una inserción. La frase dice entonces: El fma tca tat eth eho tdo. Esta frase claramente tiene otro significado, es decir, contiene información de sentido erróneo. De igual manera, dejar fuera la letra a en grasa también provoca una mutación de desplazamiento de marco, debido a una deleción. La frase dice entonces: El ftc ata tet je otd og. Nuevamente, esta frase claramente tiene otro significado, es decir, contiene información de sentido erróneo. Este ejemplo sugiere que las consecuencias son más dramáticas para una mutación de desplazamiento de marco que para una sustitución de pares de bases. ¡Por favor, ten en cuenta que la sustitución de un gato por un auto también puede tener enormes consecuencias en la vida diaria! |
Compuestos mutagénicos
Las sustituciones de pares de bases a menudo son causadas por sustancias electrofílicas que quieren tomar un electrón de especialmente la base de guanina nucleofílica que quiere donar un electrón para formar un par de electrones. El consecuente producto de adición de guanina (aducto) forma un par de bases con timina provocando una sustitución de pares de bases de G-C a A-T. Alternativamente, el aducto de guanina puede separarse del esqueleto fosfato-azúcar del ADN, dejando una mancha nucleotídica “vacía” en el triplete que puede ser tomada por cualquier nucleótido durante la replicación del ADN. Alternativamente, las sustituciones de pares de bases pueden ser causadas por especies reactivas de oxígeno (ROS), que son compuestos radicales que también toman un electrón de guanina y forman productos de oxidación de guanina (por ejemplo aductos de hidroxilo). Debe darse cuenta que un aducto de ADN solo puede causar un error en el orden de los nucleótidos (es decir, una mutación) si está presente durante la replicación del ADN. Antes de que una célula entre en la fase de síntesis de ADN del ciclo celular, sin embargo, el ADN se verifica minuciosamente, y los posibles errores son reparados por sistemas de reparación de ADN.
La exposición a agentes electrofílicos mutagénicos directos rara vez ocurre debido a que estas sustancias son tan reactivas que se unen inmediatamente a proteínas y ADN en nuestro alimento y medio ambiente. Por lo tanto, el daño al ADN por tales sustancias en la mayoría de los casos se origina a partir de compuestos mutagénicos indirectos, que se activan en agentes de unión al ADN durante la Fase I de la biotransformación. Este proceso de bioactivación es un efecto secundario de la biotransformación, que en realidad está apuntando a una rápida desintoxicación y eliminación de compuestos tóxicos.
Las mutaciones de desplazamiento de marco a menudo son causadas por agentes intercalantes. A diferencia de los agentes electrofílicos y ROS, los agentes intercalantes no forman enlaces covalentes con las bases de ADN. En cambio, debido a su estructura plana, los agentes intercalantes encajan exactamente entre dos nucleótidos adyacentes en la hélice del ADN. Como consecuencia, dificultan la replicación del ADN, provocando la inserción de un nucleótido extra o la deleción de un nucleótido original en la cepa de ADN replicado.
|
Prueba de Ames para mutagenicidad La mutagenicidad de un compuesto se puede probar en la prueba de Ames, que lleva el nombre de Bruce Ames, quien desarrolló el ensayo a principios de la década de 1970. El ensayo hace uso de una cepa de bacterias Salmonella que contiene una mutación en un gen que codifica una enzima involucrada en la síntesis del aminoácido histidina. En consecuencia, las bacterias ya no pueden producir histidina (convertirse en “his‑”) y volverse auxotróficas, es decir, dependen de su medio de cultivo para histidina. En el ensayo, las bacterias se exponen al compuesto de prueba en un medio que no contiene histidina. Si el compuesto de prueba no es mutagénico, la bacteria no puede crecer y morirá. Si el compuesto de prueba es mutagénico, puede provocar una retromutación (reversión) de la mutación original en unas pocas bacterias, restaurando la capacidad autotrófica de la bacteria (es decir, su capacidad para producir su propia histidina). El crecimiento de bacterias mutadas en el medio empobrecido en histidina puede ser seguido por el recuento de colonias (en una placa de agar) o por la medición de la actividad metabólica (en un ensayo de fluctuación). Los compuestos mutagénicos directos se pueden probar en la prueba de Ames sin tratamiento adicional. Sin embargo, los compuestos mutagénicos indirectos tienen que ser bioactivados antes de ejercer su acción mutagénica. Para ello, se agrega un homogeneizado de hígado al medio de cultivo que contiene todas las enzimas y cofactores requeridos para la biotransformación de Fase I del compuesto de prueba. Este homogeneizado hepático con actividad inducida del citocromo P450 (cyp) se obtiene generalmente de ratas expuestas a inductores de tipo mixto (es decir, cyp1a, cyp2b, cyp3a), como la mezcla de PCB Aroclor 1254. |
Compuestos implicados en la promoción y progresión tumoral
Como se indicó anteriormente, los carcinógenos no mutagénicos están involucrados en la estimulación de la promoción tumoral. Las sustancias promotoras de tumores estimulan la proliferación celular e inhiben la diferenciación celular y apoptosis. A diferencia de los compuestos mutagénicos, los compuestos promotores de tumores no interfieren directamente con el ADN y su efecto es reversible. Muchas sustancias endógenas (por ejemplo, hormonas) pueden actuar como agentes promotores de tumores.
| La primera ilustración de que los químicos pueden inducir cáncer proviene del caso de los deshollinadores en Londres alrededor de 1775. El cirujano Percival Pott (1714-1788) notó que muchos pacientes varones adolescentes que habían desarrollado cáncer de escroto habían trabajado durante su infancia como barredora de chimeneas. Pott estableció un vínculo directo entre la exposición al hollín durante la infancia y el desarrollo del cáncer a una edad posterior. Con base en este descubrimiento, tomar una ducha después del trabajo se volvió obligatorio para los niños que trabajaban como deshollinadores, y la incidencia observada de cáncer de escroto disminuyó. Como tal, Percival Pott fue la primera persona (i) en vincular el desarrollo del cáncer con sustancias químicas, (ii) vincular la exposición temprana con el desarrollo posterior del cáncer, y (iii) en obtener una mejor salud ocupacional al disminuir la exposición a través de una mejor higiene. En retrospectiva, ahora sabemos que los mutágenos involucrados fueron hidrocarburos aromáticos policíclicos (HAP) que fueron bioactivados en metabolitos diol-epóxido altamente reactivos. El retraso en el desarrollo del cáncer después de la exposición en la primera infancia puede atribuirse a la ausencia de un promotor tumoral. Sólo después de que los deshollinadores habían pasado por la pubertad tuvieron niveles suficientes de testosterona, lo que estimula el crecimiento del tejido escroto y en este caso actuó como agente endógeno promotor de tumores. |
La progresión tumoral es el resultado de una actividad transcripcional aberrante de alteraciones genéticas o epigenéticas. Las alteraciones genéticas pueden ser causadas por sustancias que dañan el ADN (llamadas sustancias genotóxicas) y con ello introducen roturas de hebra y división cromosómica incorrecta después de la mitosis. Esto da como resultado las características cromosómicas inestables típicas de una célula tumoral maligna, es decir, un cariotipo que consiste en un número reducido y aumentado de cromosomas (llamados aneuploidía y poliploidía, respectivamente) y estructuras cromosómicas dañadas (abberaciones). Las sustancias químicas que causan aneuploidía se llaman aneugenos y las sustancias que causan abberaciones cromosómicas se llaman clastógenos. Las sustancias genotóxicas también son muy a menudo compuestos mutagénicos. Múltiples mutaciones en los llamados protooncogenes y genes supresores de tumores son necesarias para transformar una célula normal en una célula tumoral. En una célula sana, la proliferación celular está bajo control por protooncogenes que estimulan la proliferación celular y genes supresores de tumores que inhiben la proliferación celular. En una célula cancerosa se altera el equilibrio entre protooncogenes y genes supresores de tumores: los protooncogenes actúan como oncogenes, lo que significa que estimulan continuamente la proliferación celular, debido a mutaciones y poliploidía, mientras que los genes supresores de tumores se han vuelto inactivos debido a mutaciones y aneuploidía.
Las alteraciones epigenéticas son cambios en el ADN, pero no en su orden de nucleótidos. Los cambios epigenéticos típicos incluyen cambios en la metilación del ADN, modificaciones de histonas y expresión de microARN. Los compuestos que cambian el epigenoma pueden estimular la progresión tumoral, por ejemplo, estimulando la expresión de oncogenes e inhibiendo la expresión de genes supresores de tumores. El papel en la promoción y progresión tumoral de sustancias capaces de inducir cambios epigenéticos es un campo de estudio continuo.
¿Cuáles son las tres características de las diferentes etapas de desarrollo del cáncer?
¿Cuál es la diferencia entre una sustancia mutagénica directa y una indirecta?
¿Explicar el principio de la prueba de Ames?
¿Cuál es la diferencia entre una sustitución de pares de bases y una mutación de desplazamiento de marco?
4.2.8. Alteración endocrina
Autor: Majorie van Duursen
Revisor: Timo Hamers, Andreas Kortenkamp
Objetivos de aprendizaje
Deberías ser capaz de
- explicar cómo los xenobióticos pueden interactuar con el sistema endocrino y las acciones hormonales;
- describir el sistema tiroideo y los objetivos moleculares para la alteración de la hormona tiroidea
- explicar el concepto “es el momento de la dosis lo que hace el veneno”.
Palabras clave: Sistema endocrino; Químico Disruptor Endocrino (EDC); DES; Interrupción hormonal tiroidea; Efectos multi y transgeneracionales
Breve historia
El sistema endocrino juega un papel esencial en la regulación a corto y largo plazo de una variedad de procesos bioquímicos y fisiológicos, como el comportamiento, la reproducción, el crecimiento así como los aspectos nutricionales, la función intestinal, cardiovascular y renal y la respuesta al estrés. Como consecuencia, las sustancias químicas que causan cambios en la secreción hormonal o en la actividad de los receptores hormonales pueden dirigirse a muchos órganos y funciones diferentes y pueden provocar trastornos del sistema endocrino y efectos adversos para la salud. La naturaleza y el tamaño de los efectos endocrinos causados por los químicos dependen del tipo de sustancia química, el nivel y la duración de la exposición, así como del momento de la exposición.
El “desastre de drogas DES” es uno de los ejemplos más llamativos de que los químicos endocrinos activos pueden tener graves efectos adversos para la salud en humanos. Hubo un tiempo en que el estrógeno sintético dietilestilbestrol (DES) se consideró un fármaco milagroso (Figura 1). El DES se prescribió entre los años 1940 y 1970 a millones de mujeres en todo el mundo para prevenir abortos espontáneos, abortos y trabajos de parto prematuros. Sin embargo, a principios de la década de 1970 se encontró que las hijas de madres que tomaron DES durante su embarazo tienen un mayor riesgo de desarrollar un tipo específico de cáncer vaginal y cervical. Otros estudios posteriores demostraron que las mujeres que habían estado expuestas al DES en el útero (en el útero) también tenían otros problemas de salud, como aumento del riesgo de cáncer de mama, mayor incidencia de malformaciones genitales, infertilidad, abortos espontáneos y embarazos complicados. Ahora, incluso dos generaciones después, nacen bebés con malformaciones del tracto reproductivo que se sospecha que son causadas por esta droga que sus bisabuelas tomaron durante el embarazo. Los efectos del DES se atribuyen al hecho de que es un estrógeno sintético (es decir, un compuesto xenobiótico que tiene propiedades similares al estrógeno natural 17β-estradiol), alterando así la regulación endocrina normal así como los procesos epigenéticos durante el desarrollo (enlace a la sección sobre Desarrollo Toxicidad).
Alrededor de la misma época del desastre de las drogas del DES, Rachel Carson escribió un best-seller del New York Times llamado Silent Spring. El libro se centró en las propiedades disruptivas endocrinas de contaminantes ambientales persistentes, como el insecticida DDT (Dicloro difenil tricloroetano). Ella escribió que estos contaminantes ambientales eran muy degradables en el ambiente y causan fallas reproductivas y disminución de la población en una variedad de vida silvestre. En el momento en que se publicó el libro, la disrupción endocrina era una teoría científica controvertida que se encontró con mucho escepticismo ya que la evidencia empírica carecía en gran medida. Aún así, el libro de Rachel Carson ha fomentado discusiones científicas, sociales y políticas sobre la disrupción endocrina. En 1996 se publicó otro libro científico popular que presentó más evidencia científica para advertir contra los efectos de la disrupción endocrina: Nuestro futuro robado: ¿Estamos amenazando nuestra fertilidad, inteligencia y supervivencia? Una historia de detectives científicos de Theo Colborn, Dianne Dumanoski y John Peterson Myers.

Actualmente, la alteración endocrina es un concepto ampliamente aceptado y muchos estudios científicos han demostrado una amplia variedad de efectos adversos para la salud que se atribuyen a la exposición a compuestos activos endocrinos en nuestro medio ambiente. Los estudios epidemiológicos en humanos han mostrado aumentos dramáticos en la incidencia de enfermedades relacionadas con hormonas, como cáncer de mama, ovario, testículo y próstata, enfermedades endometriales, infertilidad, disminución de la calidad espermática y enfermedades metabólicas. Considerando que las hormonas juegan un papel destacado en la aparición de estas enfermedades, es muy probable que la exposición a compuestos alteradores endocrinos contribuya a estas incidencias incrementadas de enfermedades en humanos. En la vida silvestre, los efectos de la alteración endocrina incluyen efectos feminizantes y desmasculinizantes que conducen a comportamientos sexuales desviados y fallas reproductivas en muchas especies, como peces, ranas, aves y panteras. Un ejemplo llamativo de alteración endocrina se puede encontrar en la población de caimanes del lago Apopka. El lago Apopka es el tercer lago más grande del estado de Florida, ubicado a pocos kilómetros al noroeste de Orlando. En julio de 1980, las fuertes lluvias provocaron el derrame de enormes cantidades de DDT en el lago por parte de un fabricante local de plaguicidas. Después de eso, la población de caimanes en el lago Apopka comenzó a mostrar un declive dramático. Tras un examen más detenido, estos caimanes presentaron mayores niveles de estradiol y menor testosterona en su sangre, lo que provocó testículos poco desarrollados y penes extremadamente pequeños en la descendencia masculina y ovarios severamente malformados en las hembras.
Qué hay en un nombre: definición EDC
Desde las primeras discusiones sobre la alteración endocrina, la Organización Mundial de la Salud (OMS) ha publicado varios informes para presentar el estado de la técnica en evidencia científica sobre la alteración endocrina, los efectos adversos asociados a la salud y los mecanismos subyacentes. En 2002, la OMS propuso una definición para un compuesto disruptor endocrino (EDC), que aún se está utilizando. Según la OMS, un EDC puede definirse como “una sustancia o mezcla exógena que altera la función o funciones del sistema endocrino y en consecuencia causa efectos adversos para la salud en un organismo intacto, o su progenie, o (sub) poblaciones”. En 2012, la OMS afirmó que “las EDC tienen la capacidad de interferir con el desarrollo y la función de los tejidos y órganos, y por lo tanto pueden alterar la susceptibilidad a diferentes tipos de enfermedades a lo largo de la vida. Esta es una amenaza global que hay que resolver”. La Agencia Europea del Medio Ambiente concluyó en 2012 que “la alteración endocrina inducida químicamente probablemente afecte la salud endocrina humana y silvestre en todo el mundo”. Un informe reciente (Demeneix & Slama, 2019) que fue encargado por el Parlamento Europeo concluyó que la falta de consideración EDC en los procedimientos regulatorios es “claramente perjudicial para el medio ambiente, la salud humana, la sociedad, la sostenibilidad y muy probablemente para nuestra economía”.
El sistema endocrino
Los animales superiores, incluidos los humanos, han desarrollado un sistema endocrino que les permite regular su entorno interno. El sistema endocrino está interconectado y se comunica bidireccionalmente con los sistemas neurológicos e inmunológicos. El sistema endocrino consiste en glándulas que secretan hormonas, las propias hormonas y objetivos que responden a la hormona. Las glándulas que secretan hormonas incluyen la hipófisis, tiroides, glándulas suprarrenales, gónadas y páncreas. Hay tres clases principales de hormonas: hormonas derivadas de aminoácidos (por ejemplo, hormonas tiroideas T3 y T4), hormonas peptídicas (por ejemplo, hormonas pancreáticas) y hormonas esteroides (por ejemplo, testosterona y estradiol). Las hormonas provocan una amplia variedad de respuestas biológicas, que casi siempre comienzan con la unión de una hormona a un receptor en su tejido diana. Esto desencadenará una cadena de eventos intracelulares y eventualmente una respuesta fisiológica. Comprender las características químicas de una hormona y su función, puede ayudar a explicar los mecanismos por los cuales los químicos pueden interactuar con el sistema endocrino y posteriormente causar efectos adversos para la salud.
Mecanismo de acción
Inherente a la naturaleza compleja del sistema endocrino, la alteración endocrina viene en muchas formas y formas. Puede ocurrir a nivel de receptor (enlace a la sección sobre Interacción del Receptor), pero los disruptores endocrinos también pueden perturbar la síntesis, metabolismo o transporte de hormonas (localmente o en todo el cuerpo), o mostrar una combinación de múltiples mecanismos. Por ejemplo, el DDT puede disminuir los niveles de testosterona a través del aumento de la conversión de testosterona por la enzima aromatasa, pero también actúa como un antiandrógeno bloqueando el receptor de andrógenos y como estrógeno al activar el receptor de estrógeno. Los PCB, bifenilos policlorados, son sustancias químicas que alteran la hormona tiroidea bien caracterizadas. Los PCB son químicos industriales que fueron ampliamente utilizados en los transformadores hasta su prohibición en la década de 1970, pero, debido a su persistencia, los PCB aún se pueden encontrar ubicuamente en el medio ambiente, muestras de sangre y tejidos humanos y de vida silvestre (enlace a la sección sobre COP). Se sabe que los PCB interfieren con el sistema tiroideo mediante la inhibición de la síntesis de la hormona tiroidea y/o aumentando el metabolismo de la hormona tiroidea, inhibiendo la unión de las hormonas tiroideas a las proteínas de unión al suero o bloqueando la capacidad de las hormonas tiroideas Estos efectos disruptivos tiroideos pueden ocurrir en diferentes órganos a lo largo del cuerpo (ver Figura 1 en Gilbert et al., 2012).
El concepto de dosis
En el siglo XVIII, el médico y alquimista Paracelso enunció el paradigma toxicológico: “Todo es un veneno. Sólo la dosis hace que algo no sea un veneno” (enlace a la sección Relaciones concentración-respuesta, y a Introducción). Generalmente, esto se entiende como “el efecto del veneno aumenta con la dosis”. De acuerdo con este paradigma, al determinar los niveles de exposición donde comienza y termina la respuesta tóxica, se pueden derivar niveles de seguridad para proteger a humanos, animales y su entorno. Sin embargo, la interpretación e implementación práctica de este concepto se ve desafiada por problemas que han surgido en la toxicología moderna, especialmente con las EDC, como las curvas dosis-respuesta no monótonas y el tiempo de exposición.
Para establecer una relación dosis-respuesta, tradicionalmente se realizan experimentos toxicológicos donde animales adultos están expuestos a dosis muy altas de un químico. Para determinar un nivel seguro, se determina la dosis de prueba más alta a la que no se observa ningún efecto tóxico (el nivel de NOAEL o ningún efecto adverso observado) y se agrega un factor adicional de “seguridad” o “incertidumbre” de generalmente 100. Este factor 100 explica las diferencias entre animales experimentales y humanos, y las diferencias dentro de la población humana (ver capítulo 6 sobre Evaluación de riesgos). Las exposiciones por debajo del nivel de seguridad generalmente se consideran seguras. Sin embargo, en los últimos años, los estudios que miden los efectos de los químicos hormonalmente activos también comenzaron a mostrar los efectos biológicos de los químicos activos endocrinos a concentraciones extremadamente bajas, que se presumieron que eran seguras y están en el rango de niveles de exposición humana. Existen varias explicaciones fisiológicas a este fenómeno. Es importante darse cuenta de que las respuestas hormonales endógenas no actúan de manera lineal, monotónica (i.c. el efecto va en una dirección), como se puede observar en la Figura 2 para los niveles de hormona tiroidea y el coeficiente intelectual. Existen bucles de retroalimentación para regular el sistema endocrino en caso de sobreestimulación o subestimulación de un receptor y hay claras diferencias tisulares en la expresión del receptor y la sensibilidad a las acciones hormonales. Además, las hormonas son mensajeros, que están diseñados para transferir un mensaje a través del cuerpo. Hacen esto a concentraciones extremadamente bajas y pequeños cambios en las concentraciones hormonales pueden causar grandes cambios en la ocupación del receptor y la actividad del receptor. A altas concentraciones, el cambio en la ocupación del receptor es solo mínimo. Esto significa que los efectos a dosis altas no siempre predicen los efectos de las EDC a dosis más bajas y viceversa.
Cada vez es más claro que no solo la dosis, sino también el momento de la exposición juega un papel importante en la determinación de los efectos de las EDC en la salud. Estudios multigeneracionales muestran que la exposición a EDC en el útero puede afectar a las generaciones futuras (Figura 3). Los estudios sobre los nietos y nietas cuyas madres estuvieron expuestas prenatalmente al DES son limitados ya que apenas comienzan a llegar a la edad en que se pueden estudiar problemas de salud relevantes, como la fertilidad. Sin embargo, los estudios en roedores con DES, bisfenol-A y DEHP muestran que las madres expuestas perinatalmente tienen nietos con malformaciones del tracto reproductivo, así como una mayor susceptibilidad a tumores mamarios en descendencia femenina y cáncer testicular y mala calidad de semen en crías masculinas. Algunos estudios incluso muestran efectos en los bisnietos (generación F3), lo que indica que los efectos disruptores endocrinos se han pasado a las siguientes generaciones sin exposición directa de estas generaciones. A estos se les llama efectos transgeneracionales. Se cree que los efectos retardados a largo plazo de las EDC surgen de modificaciones epigenéticas en las células (germinales) y pueden ser irreversibles y transgeneracionales (enlace a la sección Toxicidad del Desarrollo). En consecuencia, los niveles seguros de exposición a EDC pueden variar, dependiendo del momento de la exposición. La exposición de adultos a EDC a menudo se considera activacional, por ejemplo, un compuesto similar al estrógeno como el DES puede estimular la proliferación de células mamarias sensibles al estrógeno en un adulto que conduce al cáncer de mama. Cuando la exposición a EDC ocurre durante el desarrollo, los efectos se consideran organizativos, por ejemplo, el DES cambia el desarrollo de células germinales de las madres expuestas perinatalmente y posteriormente conduce a malformaciones del tracto genital en sus nietos. Los efectos multigeneracionales son claros en los estudios con roedores, pero no son tan claros en humanos. Esto se debe a que es difícil caracterizar la exposición a EDC en generaciones anteriores (que puede durar más de 100 años en humanos), y es difícil filtrar el efecto de un EDC específico ya que los humanos están expuestos a una miríada de químicos a lo largo de sus vidas.
EDCs en el medio ambiente
Algunos ejemplos bien conocidos de EDC son pesticidas (por ejemplo, DDT), suavizantes plásticos (por ejemplo, ftalatos, como DEHP), precursores plásticos (por ejemplo, bisfenol-A), productos químicos industriales (por ejemplo, PCB), repelentes de agua y manchas (sustancias perfluoradas como PFOS y PFOA) y hormonas sintéticas (por ejemplo, DES). La exposición a los EDC puede ocurrir a través del aire, polvo doméstico, lixiviación en alimentos y piensos, desechos y agua potable. La exposición suele ser involuntaria y a bajas concentraciones, a excepción de los medicamentos hormonales. Claramente, las hormonas sintéticas también pueden tener efectos beneficiosos. Los cánceres hormonales como los cánceres de mama y próstata se pueden tratar con hormonas sintéticas. Y piensa en la píldora anticonceptiva que ha cambiado la vida de muchas mujeres en todo el mundo desde la década de 1960. Hoy en día, ningún otro método es tan ampliamente empleado en tantos países del mundo como la píldora anticonceptiva, con una estimación de 75 millones de usuarios entre las mujeres en edad reproductiva con pareja. Un desafortunado efecto secundario de esto es el aumento de los niveles hormonales de drogas en nuestro entorno que conduce a la feminización de peces machos nadando en las aguas contaminadas. Las hormonas farmacéuticas, junto con las hormonas producidas de forma natural, son excretadas por mujeres y hombres y estas no se eliminan completamente a través de tratamientos convencionales de aguas residuales. Además, varios productos farmacéuticos que no se considera que actúan a través del sistema endocrino, de hecho pueden mostrar actividad endocrina y causar fallas reproductivas en los peces. Estos son por ejemplo el betabloqueante atenolol, el fármaco antidiabético metformina y el analgésico paracetamol.
Lectura adicional:
OMS-IPCS. Evaluación global del estado de la ciencia de los disruptores endocrinos. 2002 https://www.who.int/ipcs/publications/new_issues/endocrine_disruptors/en/
OMS-PNUMA. Estado de la ciencia de los productos químicos disruptores endocrinos.2012 www.who.int/iris/bitstream/10665/78101/1/9789241505031_eng.pdf
Agencia Europea del Medio Ambiente (EEE) Los impactos de los disruptores endocrinos en la vida silvestre, las personas y sus entornos El informe Weybridge+15 (1996-2011). EEE Informe técnico No 2/2012 EEE Copenhague. ISSN 1725-2237 https://www.eea.europa.eu/publications/the-impacts-of-endocrine-disrupters
Demeneix, B., Slama, R. (2019) Disruptores endocrinos: de la evidencia científica a la protección de la salud humana. Departamento de Política de Derechos Ciudadanos y Asuntos Constitucionales Dirección General de Políticas Internas de la Unión PE 608.866 http://www.europarl.europa.eu/RegData/etudes/STUD/2019/608866/IPOL_STU(2019)608866_EN.pdf
¿Cuáles son los mecanismos posibles para reducir la acción de una determinada hormona?
Dar tres sitios diana para la discución de la hormona tiroidea y nombrar el proceso biológico en ese sitio diana que puede ser afectado por un EDC.
¿Qué mecanismo puede provocar la desmasculinización de los alineadores masculinos por DDT?
¿Por qué es importante el momento de la exposición al evaluar el riesgo de exposición a EDC?
4.2.9. Toxicidad del desarrollo
Autor (es): Jessica Legradi, Marijke de Cock
Revisor: Paul Fowler
Objetivos de aprendizaje
Deberías ser capaz de
- explicar los seis principios de la teratología
- nombrar la diferencia entre deformación, malformación y síndrome
- describir el principio de DoHad
- indican qué es la epigenética y qué mecanismos epigenéticos podrían conducir a efectos transgeneracionales
Palabras clave: Teratogenicidad, toxicidad para el desarrollo, DoHAD, Epigenética, transgeneracional
Toxicidad del desarrollo
La toxicidad del desarrollo se refiere a cualquier efecto adverso, causado por factores ambientales, que interfiere con la homeostasis, el crecimiento normal, la diferenciación o el desarrollo antes de la concepción (ya sea de los padres), durante el desarrollo prenatal o posnatalmente hasta la pubertad. Los efectos pueden ser reversibles o irreversibles. Los factores ambientales que pueden tener un impacto en el desarrollo son factores de estilo de vida como el alcohol, la dieta, el tabaquismo, las drogas, los contaminantes ambientales o factores físicos. Cualquier cosa que pueda perturbar el desarrollo del embrión o feto y produzca una malformación se llama teratógeno. Los teratógenos pueden interrumpir un embarazo o producir efectos adversos llamados malformaciones congénitas (defectos de nacimiento, anomalía). Una malformación se refiere a cualquier efecto sobre el desarrollo estructural de un feto (por ejemplo, retraso, mala dirección o detención de procesos de desarrollo). Las malformaciones ocurren principalmente temprano en el desarrollo y son permanentes. Las malformaciones no deben confundirse con deformaciones, que en su mayoría son efectos temporales causados por fuerzas mecánicas (por ejemplo, moldeo de la cabeza después del nacimiento). Un teratógeno puede inducir varias malformaciones diferentes. Todas las malformaciones causadas por un teratógeno se denominan síndrome (por ejemplo, síndrome alcohólico fetal).
Seis principios de teratología (por James G. Wilson)
En 1959 James G. Wilson publicó los 6 principios de la teratología. Hasta ahora estos principios todavía se ven como los fundamentos de la toxicología del desarrollo. Los principios son:
1. La susceptibilidad a la teratogénesis depende del genotipo del conceptus y de la manera en que éste interactúe con factores ambientales adversos
Diferencias de especies: diferentes especies pueden reaccionar diferentes (sensibilidades) al mismo teratógeno. Por ejemplo, la talidomida (ablandamiento) un medicamento que se usa para tratar las náuseas matutinas de una mujer embarazada causa malformaciones graves de las extremidades en humanos mientras que tales efectos no se observaron en ratas y ratones.
Diferencias entre cepas e intra camadas: los antecedentes genéticos de individuos dentro de una especie pueden causar diferencias en la respuesta a un teratógeno.
Interacción del genoma y el medio ambiente: organismos del mismo origen genético pueden reaccionar de manera diferente a un teratógeno en diferentes ambientes.
Causalidad multifactorial: el resumen de lo anterior. La gravedad de una malformación depende de la interacción de varios genes (inter e intra especies) y varios factores ambientales.
2. La susceptibilidad a la teratogénesis varía según la etapa de desarrollo en el momento de la exposición a una influencia adversa
Durante el desarrollo hay periodos en los que el feto es específicamente sensible a una determinada malformación (Figura 1). En general, el desarrollo muy temprano (embrionario) es más susceptible a los efectos teratogénicos.
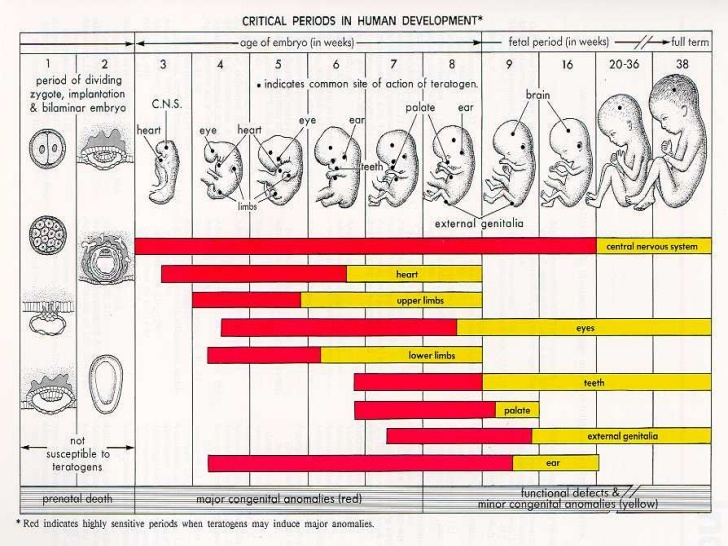
3. Los agentes teratogénicos actúan de maneras específicas (mecanismos) sobre células y tejidos en desarrollo para iniciar secuencias de eventos anormales del desarrollo (patogénesis)
Cada agente teratogénico produce un patrón distintivo de malformación. Un ejemplo es el síndrome alcohólico fetal, que induce apariencia anormal, estatura corta, bajo peso corporal, tamaño pequeño de la cabeza, mala coordinación, baja inteligencia, problemas de comportamiento, problemas de audición o visión y rasgos faciales muy característicos (aumento de la distancia entre los ojos).
4. El acceso de influencias adversas a los tejidos en desarrollo depende de la naturaleza de la influencia (agente)
Los teratógenos pueden ser radiación, infecciones o químicos, incluidos los medicamentos. El efecto teratogénico depende de la concentración de un teratógeno que llegue al embrión. La concentración en el embrión está influenciada por la absorción materna, metabolización y eliminación y el tiempo que el agente necesita para llegar al embrión. Esto puede ser muy diferente entre los teratógenos. Por ejemplo, la radiación fuerte también es un teratógeno fuerte ya que llega fácilmente a todos los tejidos del embrión. Esto también significa que un compuesto probado para ser teratogénico en una prueba in vitro con embriones en un tubo podría no ser teratogénico a un embrión en el útero de un ser humano o ratón ya que el teratógeno puede no alcanzar el embrión a una concentración crítica.
5. Las cuatro manifestaciones del desarrollo desviado son muerte, malformación, retraso del crecimiento y déficit funcional
Un teratógeno puede causar efectos menores como déficits funcionales (por ejemplo, coeficiente intelectual reducido), retrasos en el crecimiento o efectos adversos como malformaciones o muerte. Dependiendo del momento de la exposición y el grado de sensibilidad genética, un embrión tendrá mayor o menor riesgo de muerte o malformaciones. Muy temprano en el desarrollo, durante las primeras divisiones celulares, un embrión tendrá más probabilidades de morir en lugar de ser implantado y desarrollándose más.
6. Las manifestaciones de desarrollo desviado aumentan en frecuencia y grado a medida que aumenta la dosis, desde el nivel sin efecto hasta el 100% letal
El número de efectos y la gravedad de los efectos aumenta con la concentración de un teratógeno. Esto significa que hay una concentración umbral por debajo del cual no se producen efectos teratogénicos (sin concentración de efecto).
Orígenes del desarrollo de la salud y la enfermedad (DoHad)
El concepto de Origen del Desarrollo de la Salud y la Enfermedad (DoHAD) describe que los factores ambientales tempranos en la vida contribuyen a la salud y la enfermedad más adelante en la vida. La base de este concepto fue la hipótesis de Barker, la cual se formuló como una explicación del aumento de la mortalidad relacionada con enfermedades cardiovasculares (ECV) en el Reino Unido entre 1900 y 1950. Barker y sus colegas observaron que la prevalencia de ECV y accidente cerebrovascular se correlacionó con la mortalidad neonatal y posnatal (Figura 2). Esto los llevó a formular la hipótesis de que la mala nutrición temprana en la vida conduce a un mayor riesgo de enfermedad cardiovascular y accidente cerebrovascular más adelante en la vida. Posteriormente, esto se desarrolló en la hipótesis del fenotipo ahorrativo afirmando que la mala nutrición en el útero programas de mecanismos adaptativos que permiten lidiar con condiciones pobres en nutrientes en la vida posterior, pero también pueden resultar en una mayor susceptibilidad al síndrome metabólico. Esta hipótesis ahorrativa finalmente se desarrolló en la teoría DoHad.
El efecto de la nutrición temprana en la salud de los adultos se ilustra claramente en el Estudio de Cohorte de Nacimiento de Hambruna Holandesa. En esta cohorte se estudiaron retrospectivamente mujeres y hombres que nacieron durante o justo después de la hambruna holandesa. La hambruna holandesa fue una hambruna que tuvo lugar en la parte occidental de los Países Bajos ocupados por Alemania en el invierno de 1944-1945. Su duración de 3 meses crea la posibilidad de estudiar el efecto de la mala nutrición durante cada trimestre individual del embarazo. Se pueden esperar efectos sobre el peso al nacer, por ejemplo, si se restringe la ingesta calórica durante el embarazo. Esto fue, sin embargo, sólo el caso cuando la hambruna ocurrió durante el segundo o tercer trimestre. Los niveles más altos de glucosa e insulina en la edad adulta solo se observaron para los expuestos en el tercer trimestre, mientras que los expuestos durante el segundo trimestre mostraron una mayor prevalencia de enfermedad obstructiva de las vías respiratorias. Estos efectos no se observaron para los otros trimestres, lo que puede explicarse por el momento de restricción calórica durante el embarazo: durante el embarazo normal los islotes pancreáticos se desarrollan durante el tercer trimestre, mientras que durante el segundo trimestre se sabe que el número de células pulmonares se duplica.
El concepto DoHad no solo se enfoca en la nutrición temprana, sino que incluye todo tipo de factores estresantes ambientales durante el período de desarrollo que pueden contribuir a la enfermedad del adulto, incluida la exposición a compuestos químicos. Los productos químicos pueden provocar efectos como alteración endocrina o neurotoxicidad, que pueden conducir a cambios morfológicos y fisiológicos permanentes cuando ocurren temprano en la vida. Ejemplos bien conocidos de tales químicos son dietilestilbestrol (DES) y diclorodifeniltricloroetano (DDT). El DES fue una droga estrogénica que se administró a mujeres entre 1940 y 1970 para prevenir el aborto espontáneo. Se retiró del mercado en 1971 debido a los efectos cancerígenos así como un mayor riesgo de infertilidad en niños que estuvieron expuestos in útero (enlace a la sección sobre Interrupción endocrina). El DDT es un pesticida que ha sido prohibido en la mayoría de los países, pero que todavía se usa en algunos para el control de la malaria. Varios estudios, incluyendo un análisis agrupado de siete cohortes europeas, encontraron asociaciones entre los niveles de exposición al DDT in utero y el crecimiento infantil y la obesidad.
La presencia ubicua de químicos en el medio ambiente hace que sea extremadamente relevante estudiar los efectos sobre la salud en humanos, pero también lo hace muy desafiante ya que prácticamente no existe un grupo de control perfecto. Esto enfatiza la importancia de la prevención, que es el mensaje clave del concepto DoHad. El estilo de vida adulto y la exposición correspondiente a compuestos tóxicos siguen siendo factores modificables importantes tanto para el tratamiento como para la prevención de enfermedades. Sin embargo, como la plasticidad del desarrollo, y por lo tanto el potencial de cambio, es mayor en la etapa temprana de la vida, es importante enfocarse en la exposición en las fases tempranas: durante el embarazo, la infancia, la infancia y la adolescencia. Esto se refleja en los reguladores que frecuentemente imponen niveles de exposición tolerables más bajos para los bebés en comparación con los adultos.
Epigenética
Para algunos compuestos en el útero se sabe que la exposición causa efectos más adelante en la vida (ver DoHad) o incluso inducir efectos en la descendencia o nieta del embrión expuesto (efecto transgeneracional). El dietilestilbesterol (DES) es un compuesto para el cual se reportan efectos transgeneracionales. Se administró DES a mujeres embarazadas para reducir el riesgo de abortos espontáneos y otras complicaciones del embarazo. Las mujeres que tomaron DES durante el embarazo tienen un riesgo ligeramente mayor de cáncer de mama. Las hijas expuestas en el útero, por otro lado, tenían una alta tendencia a desarrollar tumores vaginales raros. En la tercera generación se observaron mayores incidencias de infertilidad, cáncer de ovario y un mayor riesgo de defectos congénitos. Sin embargo, los datos disponibles para la tercera generación son pequeños y, por lo tanto, hasta el momento solo poseen evidencia limitada. Otro compuesto sospechoso de causar efectos transgeneracionales es el fungicida vinclozolina. La vinclozolina es una sustancia química antiandrogénica que altera el sistema endocrino. Se ha demostrado que la exposición a vinclozolina conduce a efectos transgeneracionales sobre la función testicular en ratones.
Los efectos transgeneracionales pueden ser inducidos a través de alteraciones genéticas (mutaciones) en el ADN. De esta manera se altera el orden de los nucleótidos en el genoma del gametocito parental y esta alteración se hereda a la descendencia. Alternativamente, los efectos transgeneracionales pueden ser inducidos por medio de alteraciones epigenéticas. La epigenética se define como el estudio de los cambios en la expresión génica que ocurren sin cambios en la secuencia del ADN, y que son heredables en la progenie de células u organismos. Los cambios epigenéticos ocurren de forma natural pero también pueden estar influenciados por factores de estilo de vida o enfermedades o contaminantes ambientales. Las alteraciones epigenéticas son una forma especial de toxicología del desarrollo, ya que los efectos podrían no causar malformaciones teratogénicas inmediatas. En cambio, los efectos pueden ser visibles solo más adelante en la vida o en las generaciones posteriores. Se supone que los compuestos pueden inducir cambios epigenéticos y con ello provocar efectos transgeneracionales. Para DES y vinclozolina se han reportado cambios epigenéticos en ratones y estos podrían explicar los cambios transgeneracionales observados en humanos. Dos mecanismos epigenéticos principales se describen generalmente como responsables de los efectos transgeneracionales, es decir, la metilación del ADN y las modificaciones de histonas.
Metilación del ADN
La metilación del ADN es la modificación epigenética más estudiada y describe la metilación de nucleótidos de citosina en el genoma (Figura 3) por ADN metiltransferasa (DNMTs). La actividad génica generalmente depende del grado de metilación de la región promotora: si el promotor está metilado, el gen suele ser reprimido. Una peculiaridad de la metilación del ADN es que puede limpiarse y reemplazarse nuevamente durante eventos de reprogramación epigenética para establecer patrones de expresión génica específicos de células y tejidos. La reprogramación epigenética ocurre muy temprano en el desarrollo. Durante esta fase se borran y remodelan las marcas epigenéticas, como las marcas de metilación. La reprogramación epigenética es necesaria ya que los genomas maternos y paternos están marcados diferencialmente y deben ser reprogramados para asegurar un desarrollo adecuado.
Modificación de histonas
Dentro del cromosoma, el ADN está densamente empaquetado alrededor de las proteínas histonas. La transcripción génica solo puede tener lugar si se afloja el empaquetamiento de ADN alrededor de las histonas. Varios procesos de modificación de histonas están involucrados en el aflojamiento de este empaquetamiento, como la acetilación, metilación, fosforilación o ubiquitinación de las moléculas de histona (Figura 4).
Figura en preparación
Figura 4: (a) El ADN se envuelve alrededor de las moléculas de histonas. Las moléculas de histonas están dispuestas de una manera que sus colas de aminoácidos están señalando fuera del paquete. Estas colas pueden ser alteradas por ejemplo vía acetilación. (b) Si las colas están acetiladas, el ADN se empaqueta con menos fuerza y los genes pueden transcribirse. Si las colas no están acetiladas el ADN se empaqueta muy apretado y la transcripción génica se ve obstaculizada.
Referencias
Barker, D.J., Osmond, C. (1986). Mortalidad infantil, nutrición infantil y cardiopatía isquémica en Inglaterra y Gales. Lanceta 1 (8489), 1077-1081.
¿Describir la diferencia entre malformación y deformación?
¿Qué factores pueden influir en la susceptibilidad de un organismo al teratógeno?
Describir en qué se diferencia DoHad de la hipótesis de Barker?
¿Cuáles son los dos mecanismos epigenéticos los principales responsables de los efectos transgeneracionales?
¿Nombrar un compuesto teratogénico?
4.2.10. Inmunotoxicidad
Autor: Nico van den Brink
Crítica: Manuel E. Ortiz-Santaliestra
Objetivos de aprendizaje:
Deberías ser capaz de:
- Comprender la complejidad de los efectos potenciales de las sustancias químicas en el sistema inmunológico
- Explicar la diferencia entre partes innatas y adquiridas del sistema inmune
- Explicar los modos de toxicidad más importantes que pueden afectar a las células inmunitarias
Palabras clave: Toxicología inmune, patógenos, sistema inmune innato y adaptativo, enfermedad de Lyme
Introducción
El sistema inmune de los organismos es muy complejo con diferentes células y otros componentes interactuando entre sí. El sistema inmunológico tiene la función de proteger al organismo de patógenos e infecciones. Consiste en una parte innata, que es activa desde la infancia y una parte adquirida que es adaptativa a la exposición a patógenos. El sistema inmunitario puede incluir diferentes componentes dependiendo de la especie (Figura 1).
Los principales órganos que intervienen en el sistema inmunitario de los mamíferos son el bazo, el timo, la médula ósea y los ganglios linfáticos. En las aves, además de todo lo anterior, también está la bursa de Fabricius. Todos estos órganos desempeñan papeles específicos en la defensa inmune, por ejemplo, el bazo sintetiza anticuerpos y juega un papel importante en la dinámica de los monocitos; el timo es el órgano donde se desarrollan las células T mientras que en la médula ósea se producen células linfoides, que son transportadas a otros tejidos para su posterior desarrollo. La bursa de Fabricius es específica para aves y es esencial para el desarrollo de células B. La sangre es un tejido importante a considerar por su papel en el transporte de células. El sistema innato generalmente brinda la primera respuesta a infecciones y patógenos, sin embargo no es muy específico. Consta de varios tipos de células con diferentes funciones como macrófagos, neutrófilos y mastocitos. Los macrófagos y neutrófilos pueden actuar contra patógenos por fagocitosis (envolviéndose en lisosomas celulares y destrucción del patógeno). Los neutrófilos son de vida relativamente corta, actúan rápido y pueden producir un estallido respiratorio para destruir el patógeno/microbio. Esto implica una rápida producción de especies reactivas de oxígeno (ROS) que pueden destruir los patógenos. Los macrófagos generalmente tienen un lapso de vida más largo, reaccionan más lento pero más prolongado y pueden atacar a través de la producción de óxido nítrico y menos a través de ROS. Los macrófagos producen citocinas para comunicarse con otros miembros del sistema inmune, especialmente los tipos celulares del sistema adquirido. Otros miembros del sistema inmune innato son los mastocitos que pueden excretar, por ejemplo, histamina en la detección de antígenos. Las células del sistema inmune adquirido o adaptativo montan respuestas más específicas para el ataque inmune, y por lo tanto generalmente son más efectivas. Los linfocitos son las células del sistema inmune adaptativo que pueden clasificarse en linfocitos B y linfocitos T. Los linfocitos B producen anticuerpos que pueden servir como receptores de antígeno de superficie celular, esenciales en el reconocimiento de, por ejemplo, microbios. Los linfocitos B facilitan respuestas inmunes humorales (extracelulares) contra microbios extracelulares (en el tracto gastrointestinal respiratorio y en la circulación sangre/linfa). Tras el reconocimiento de un antígeno, los linfocitos B producen anticuerpos de especies que se unen al antígeno específico. Esto por un lado puede disminuir la infectividad de patógenos (por ejemplo, microbios, virus) directamente, pero también marcarlos para su reconocimiento por las células fagocíticas. Los linfocitos T son activos contra patógenos y microbios intracelulares. Una vez dentro de las células, los patógenos están fuera del alcance de los linfocitos B. Los linfocitos T pueden activar macrófagos o neutrófilos para destruir patógenos fagocitados o incluso destruir células infectadas. Tanto los linfocitos B como los T son capaces de producir una diversidad extrema de clones, específicos para el reconocimiento de antígenos. La comunicación entre las diferentes células inmunitarias se produce por la producción de, por ejemplo, citocinas, incluyendo interleucinas (IL), quimiocinas, interferones (IF) y también Factores de Necrosis Tumoral (TNF). Las citocinas y los TNF están relacionados con respuestas específicas en el sistema inmune, por ejemplo, IL6 está involucrada en la activación de las células B para producir inmunoglobulinas, mientras que el TNF-α está involucrado en el inicio temprano de la inflamación, por lo tanto una de las citocinas que induce respuestas inmunes agudas. La inflamación es una respuesta genérica a patógenos montados por células de la parte innata del sistema inmune. Generalmente se traduce en un aumento de la temperatura e hinchazón del tejido afectado, causado por la infiltración del tejido por leucocitos y otras células del sistema innato. Una respuesta inflamatoria aguda adecuada no solo es esencial como primera defensa sino que también facilitará la activación del sistema inmune adaptativo. La comunicación entre las células inmunes, a través de citoquinas, no solo dirige las células al lugar de infección sino que también activa, por ejemplo, las células del sistema inmunitario adquirido. Esta es una descripción muy breve y no exhaustiva del sistema inmune, para más detalles sobre el funcionamiento del sistema inmune ver por ejemplo Abbas et al. (2018).
Los químicos pueden afectar el sistema inmunológico de diferentes maneras. La exposición al plomo, por ejemplo, puede resultar en inmunosupresión en aves acuáticas y rapaces (Fairbrother et al. 2004, Vallverdú-Coll et al., 2019). La disminución del peso del bazo, el menor número de glóbulos blancos y la capacidad reducida para montar una respuesta humoral contra un antígeno específico (por ejemplo, glóbulos rojos de oveja), indicaron un menor potencial de aves expuestas para generar respuestas inmunitarias adecuadas tras la infección. La exposición al mercurio resultó en una disminución de la proliferación de células B en pinzones cebra (Taeniopygia guttata), afectando la parte adquirida del sistema inmune (Lewis et al., 2013). Sin embargo, el aumento del sistema inmune tras la exposición a, por ejemplo, cadmio también se ha reportado, por ejemplo, en pequeños mamíferos, lo que indica una mejora de la respuesta inmune (Demenesku et al., 2014). Tanto la inmunosupresión como la potenciación inmunológica pueden tener impactos negativos en los organismos involucrados; la primera puede disminuir la capacidad del organismo para tratar patógenos u otras infecciones, mientras que la potenciación inmune puede aumentar las demandas de energía del organismo y también puede resultar en, por ejemplo hipersensibilidad o incluso autoinmunidad en organismos.
Los químicos pueden afectar a las células inmunitarias a través de la toxicidad a mecanismos que no son específicos del sistema inmunitario. Dado que muchos tipos de células diferentes están involucrados en el sistema inmune, la sensibilidad a estos modos de toxicidad puede variar considerablemente entre las células y entre los químicos. Esto implicaría que en su conjunto, el sistema inmune puede incluir inherentemente células que son sensibles a diferentes químicos, y como tal puede ser bastante sensible a una variedad de tóxicos. Por ejemplo, la inducción de la apoptosis, la muerte celular programada, es esencial para eliminar las células activadas involucradas en una respuesta inmune después de minimizar la infección y el sistema está volviendo a un estado de homeostasis (ver Figura 2). Los químicos pueden inducir apoptosis, y así interferir con la cinética de las respuestas inmunes adaptativas, reduciendo potencialmente la longevidad de las células.
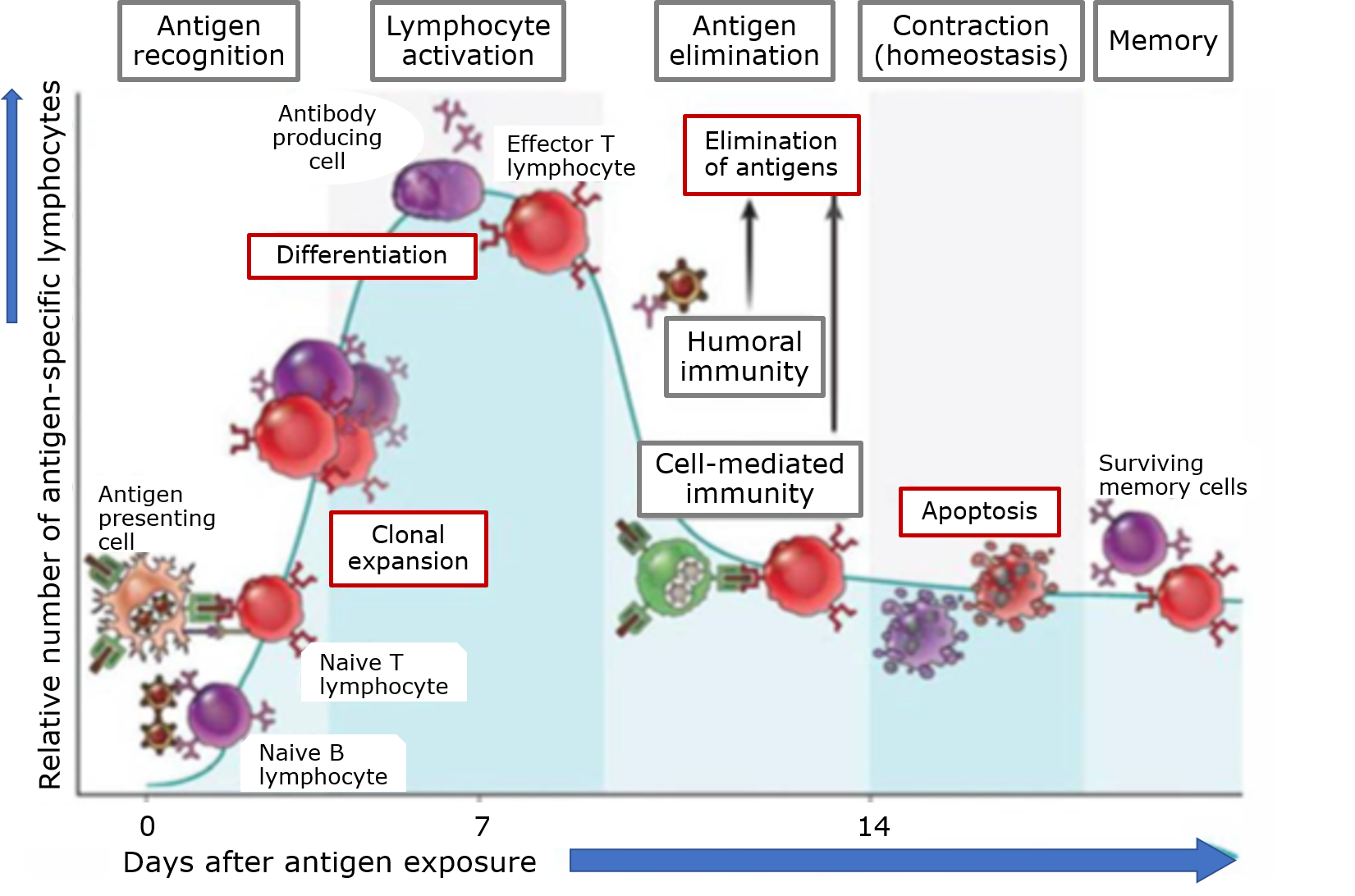
Los efectos tóxicos sobre mecanismos específicos del sistema inmune pueden estar relacionados con su funcionamiento. Dado que la producción de ROS y óxidos nítricos son vías efectoras a lo largo de las cuales neutrófilos y macrófagos de los sistemas innatos combaten patógenos (a través de una alta producción de especies reactivas de oxígeno, es decir, estallido oxidativo, para atacar patógenos), los impactos en el estado oxidativo de estas células pueden no solo resultan en toxicidad general, afectando potencialmente a una variedad de tipos celulares, pero también puede afectar la capacidad de respuesta del sistema inmune (innato) particularmente. Por ejemplo, el cadmio tiene una alta afinidad para unirse al glutatión (GSH), un antioxidante prominente en las células, y ha demostrado afectar las respuestas inmunes agudas en el timo y los bazos de ratones (Pathak y Khandelwal, 2007) a través de este mecanismo. Una disminución de GSH por la unión de químicos (como el cadmio) puede modular los macrófagos hacia una respuesta proinflamatoria por cambios en el estado redox de las células involucradas, cambiando no solo sus actividades contra patógenos sino potencialmente también su producción y liberación de citocinas (Dong et al., 1998).
GSH también está implicada en la modulación del sistema inmune adquirido al afectar a las llamadas células presentadoras de antígeno (APCs, por ejemplo, células dendríticas). Las APCs capturan antígenos microbianos que ingresan al cuerpo, los transportan a tejidos inmuno-activos específicos (por ejemplo, ganglios linfáticos) y los presentan a linfocitos T vívidos, induciendo una respuesta inmune adecuada, las llamadas células T colaboradoras. Las células T colaboradoras incluyen subconjuntos, por ejemplo, células T cooperadoras 1 (células Th1) y células T cooperadoras 2 (células Th2). Las respuestas Th1 son importantes en la defensa contra infecciones intracelulares, por activación de macrófagos para ingerir microbios. Las respuestas Th2 pueden ser iniciadas por infecciones por organismos demasiado grandes para ser fagocitados, y mediadas por, por ejemplo, alérgenos. Como se mencionó, el agotamiento de GSH puede resultar en cambios en la producción de citocinas por APC (Dong et al., 1998), afectando generalmente la liberación de citocinas promotoras de respuesta Th1. Por lo tanto, la exposición a sustancias químicas que interfieren con la cinética de GSH puede resultar en un desequilibrio entre las respuestas Th1 y Th2 y, como tal, afectar la capacidad de respuesta del sistema inmune. El cadmio y otros metales tienen una alta afinidad para unirse a GSH y, por lo tanto, pueden reducir las respuestas Th1, mientras que en contraste, los productos químicos promotores de GSH pueden reducir la capacidad de los organismos para iniciar respuestas Th2 (Pathak y Khandelwal, 2008).
La visión general sobre los posibles efectos que los químicos pueden tener sobre el sistema inmunológico tal como se presenta aquí no es exhaustiva en absoluto. Esto es aún más complicado porque los efectos pueden ser contextuales, lo que significa que los químicos pueden tener diferentes impactos dependiendo de la situación en la que se encuentre un organismo. Por ejemplo, la magnitud de los efectos inmunotóxicos puede depender del estado general del organismo, y por lo tanto algunos animales infectados pueden mostrar efectos por exposición química mientras que otros pueden no. Los impactos también pueden diferir entre los tipos de infección (por ejemplo, infecciones sensibles a Th1 versus Th2). Esto, junto con la composición compleja y dinámica del sistema inmune, limita el desarrollo de relaciones generales de respuesta a la dosis y predicciones de peligros para los químicos. Además, la mayor parte de la investigación sobre los efectos de los químicos en el sistema inmune se centra en humanos, basada en estudios en ratas y ratones. Poco se sabe sobre las diferencias entre especies, especialmente en especies no mamíferas que pueden tener sistemas inmunes completamente estructurados diferencialmente. Algunos estudios sobre fauna silvestre han mostrado efectos de metales traza en pequeños mamíferos (Tersago et al., 2004, Rogival et al., 2006, Tête et al., 2015) y del plomo en aves (Vallverdú-Coll et al., 2015). Sin embargo, los modos de acción específicos aún están por resolverse en condiciones de campo. Sin embargo, la investigación sobre la inmunotoxicidad en la vida silvestre es esencial no solo desde el punto de vista conservador (para proteger los organismos y especies involucradas) sino también desde la perspectiva de la salud humana. La vida silvestre juega un papel importante en la cinética de las enfermedades zoonóticas, por ejemplo, los pequeños mamíferos son el principal reservorio de espiroquetas de Borrelia, los patógenos causantes de la enfermedad de Lyme mientras que las aves acuáticas migratorias están indicados para impulsar la propagación de, por ejemplo, la influenza aviar. Por lo tanto, es eminente el papel de la vida silvestre en la cinética de la propagación ambiental de las enfermedades zoonóticas, las cuales pueden verse seriamente afectadas por alteraciones químicas inducidas de su sistema inmunológico.
Referencias y lecturas adicionales
Abbas, A.K., Lichtman, A.H., Pillai, S. (2018). Inmunología Celular y Molecular. 9ª Edición. Elsevier, Filadelfia, EUA. ISBN: 978-0-323-52324-0
Demenesku, J., Mirkov, I., Ninkov, M., Popov Aleksandrov, A., Zolotarevski, L., Kataranovski, D., Kataranovski, M. (2014). La administración aguda de cadmio a ratas ejerce efectos tanto inmunosupresores como proinflamatorios en el bazo. Toxicología 326, 96-108.
Dong, W., Simeonova, P.P., Gallucci, R., Matheson, J., Flood, L., Wang, S., Hubbs, A., Lustre, M.I. (1998). Los metales tóxicos estimulan citocinas inflamatorias en hepatocitos a través de mecanismos de estrés oxidativo Toxicología y Farmacología Aplicada 151, 359-366.
Fairbrother, A., Smits, J., Grasman, K.A. (2004). Inmunotoxicología aviar. Revista de Toxicología y Salud Ambiental, Parte B 7, 105-137.
Galloway, T., Handy, R. (2003). Inmunotoxicidad de plaguicidas organofosforados. Ecotoxicología 12, 345-363.
Lewis, C.A., Cristol, D.A., Swaddle, J.P., Varian-Ramos, C.W., Zwollo, P. (2013). Disminución de la respuesta inmune en Pinzones cebra expuestos a dosis subletales de mercurio. Archivos de Contaminación Ambiental y Toxicología 64, 327-336.
Pathak, N., Khandelwal, S. (2007). Papel del estrés oxidativo y la apoptosis en la atrofia tímica inducida por cadmio y esplenomegalia en ratones. Cartas de Toxicología 169, 95-108.
Pathak, N., Khandelwal, S. (2008). Impacto del cadmio en subconjuntos de linfocitos T y expresión de citocinas: regulación diferencial por estrés oxidativo y apoptosis. Biometales 21, 179-187.
Rogival, D., Scheirs, J., De Coen, W., Verhagen, R., Blust, R. (2006). Niveles sanguíneos metálicos y características hematológicas en ratones de madera (Apodemus sylvaticus L.) a lo largo de un gradiente de contaminación metálica. Toxicología y Química Ambiental 25, 149-157.
Tersago, K., De Coen, W., Scheirs, J., Vermeulen, K., Blust, R., Van Bockstaele, D., Verhagen, R. (2004). Inmunotoxicología en ratones de madera a lo largo de gradiente de contaminación por metales pesados. Contaminación Ambiental 132, 385-394.
Tête, N., Afonso, E., Bouguerra, G., Scheifler, R. (2015). Parámetros sanguíneos como biomarcadores de exposición al cadmio y plomo y efectos en ratones de madera silvestre (Apodemus sylvaticus) que viven a lo largo de un gradiente de contaminación. Quimosfera 138, 940-946.
Vallverdú-Coll, N., López-Antia, A., Martínez-Haro, M., Ortiz-Santaliestra, M.E., Mateo, R. (2015). Respuesta inmune alterada en patitos ánades reales expuestos al plomo a través de la transferencia materna en la naturaleza. Contaminación Ambiental 205, 350-356.
Vallverdú-Coll, N., Mateo, R., Mougeot, F., Ortiz-Santaliestra, M.E. (2019). Efectos inmunotóxicos del plomo en las aves. Ciencia del Medio Ambiente Total 689, 505-515.
Nombrar dos razones generales por las que la inmunomodulación en organismos puede ser muy sensible a la exposición a productos químicos ambientales
¿Por qué es que los químicos inmunomoduladores a los que los humanos no están expuestos todavía pueden tener un impacto en la salud humana?
4.2.11. Mecanismos de toxicidad de los metales
Autor: Nico M. van Straalen
Revisores: Philip S. Rainbow, Henk Schat
Objetivos de aprendizaje
Deberías ser capaz de
- enumerar cinco categorías bioquímicas de mecanismos de toxicidad metálica y describir un ejemplo para cada caso
- interpretar los síntomas bioquímicos de la toxicidad del metal (por ejemplo, categorías funcionales de los perfiles de expresión génica) y explicarlos en términos del modo de acción de un metal en particular
Palabras clave: Especies reactivas de oxígeno, unión a proteínas, unión a ADN, bombas de iones,
Sinopsis
La toxicidad de los metales a nivel bioquímico se debe a una amplia variedad de mecanismos, los cuales pueden clasificarse de la siguiente manera, aunque no son mutuamente excluyentes: (1) generación de especies radicales de oxígeno (Fe, Cu), (2) unión a grupos nucleofílicos en proteínas (Cd, Pb), (3) unión a ADN (Cr, Cd), (4) unión a canales iónicos o bombas de membrana (Pb, Cd), (5) interacción con la función de restos celulares esenciales como fosfato, grupos sulfhidrilo, hierro o calcio (As, Cd, Al, Pb). Además, estos mecanismos pueden actuar simultáneamente e interactuar entre sí. Existen patrones interesantes de susceptibilidad a los metales, por ejemplo, los mamíferos apenas son susceptibles al zinc, mientras que las plantas y los crustáceos lo son. Las lombrices, los gasterópodos y los hongos son bastante sensibles al cobre, pero no así para los vertebrados terrestres. En esta sección se discuten cinco categorías diferentes de toxicidad de metales, así como algunos patrones de diferencias de especies en la sensibilidad a los metales.
Generación de especies reactivas de oxígeno
Las especies reactivas de oxígeno (ROS) son formas activadas de oxígeno que tienen uno o más electrones desapareados en la órbita exterior. Los mejores conocidos son anión superóxido (O 2 -), oxígeno singlete (1δGo 2), peróxido de hidrógeno (H 2 O 2) y radical hidroxilo (OH •) (ver sección Estrés oxidativo), catalizadores efectivos de especies reactivas de oxígeno. Esto se relaciona con su capacidad para participar en reacciones redox con transferencia de un electrón. Una de las reacciones más famosas es la llamada reacción de Fenton catalizada por iones reducidos de hierro y cobre:
Fe 2+ + H 2 O 2 → Fe 3+ + OH • + OH -
Cu + + H 2 O 2 → Cu 2+ + OH • + OH -
Ambas reacciones producen el radical hidroxilo altamente reactivo (OH •), que puede desencadenar graves daños celulares por peroxidación de lípidos de membrana (ver la sección Estrés Oxidativo). Concentraciones muy bajas de iones metálicos pueden mantener esta reacción funcionando, ya que las formas reducidas de los iones metálicos se restauran mediante una segunda reacción con peróxido de hidrógeno:
Fe 3+ + H 2 O 2 → Fe 2+ + O 2 - + 2H +
Cu 2+ + H 2 O 2 → Cu + + O 2 - + 2H +
La reacción global es una degradación catalizada por metales del peróxido de hidrógeno, causando anión superóxido y radical hidroxilo como intermedios. El estrés oxidativo es uno de los mecanismos más importantes de toxicidad de los metales. Esto también se puede deducir del transcriptoma inducido por metales. El perfil de expresión génica ha demostrado que no es raro que más del 10% del genoma responda a concentraciones subletales de cadmio.
Fijación a proteínas
Varios metales tienen una gran afinidad hacia los grupos sulfhidrilo (-SH) en los residuos de cisteína de las proteínas. La unión a tales grupos puede distorsionar la estructura secundaria de una proteína en sitios donde los grupos SH se coordinan para formar puentes S-S. El grupo SH es un ejemplo típico de un nucleófilo, es decir, un grupo que dona fácilmente un par de electrones para formar un enlace químico. El grupo que acepta el par de electrones se llama electrófilo. Otro aminoácido en una proteína a la que se unen preferentemente los metales es la cadena lateral de imidazol de la histidina. Este grupo aromático heterocíclico con dos átomos de nitrógeno se acopla fácilmente en enlaces químicos con iones metálicos. De hecho, los residuos de histidina a menudo se utilizan en metaloproteínas para coordinar metales en el sitio activo y para transportar metales desde las raíces hacia arriba a través de los vasos del xilema de las plantas.
Un caso clásico de interacción metal-proteína con toxicidad posterior es el caso de la unión de plomo a la δ-aminolevulínico ácido deshidratasa (δ-AlAD). Esta es una enzima involucrada en la síntesis de la hemoglobina. Cataliza el segundo paso en la vía biosintética, la condensación de dos moléculas de ácido δ-aminolevulínico a una molécula de porfobilinógeno, que es un precursor de la porfirina, una unidad funcional que une hierro en la hemoglobina (Figura 1). La enzima tiene varios grupos sulfhidrilo que son susceptibles al plomo. En el eritrocito más del 80% del plomo está de hecho unido a la proteína δ-Alad (mucho más de lo que está unido a la hemoglobina). La inhibición de δ-ALAD conduce a una disminución de la síntesis de porfirina, hemoglobina insuficiente, pérdida de capacidad de absorción de oxígeno y eventualmente anemia.
Debido a que la inhibición de δ-ALAD por plomo se produce a niveles de exposición ya muy bajos, es un biomarcador muy bueno para la exposición al plomo. La medición de la actividad δ-ALAD en sangre se ha realizado extensivamente en trabajadores de industrias de procesamiento de metales y personas que viven en ambientes contaminados con metales. También en peces, aves y varios invertebrados (lombrices de tierra, planarias) se ha demostrado que el ensayo δ-ALAD es un biomarcador útil de la exposición al plomo. Además del plomo, se sabe que el mercurio inhibe δ-ALAD, mientras que las inhibiciones tanto por plomo como por mercurio pueden ser aliviadas en cierta medida por el zinc.
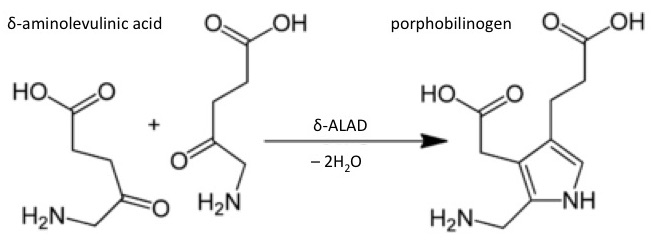
Unión de ADN
El cromo, especialmente los iones trivalentes (Cr 3+) y hexavalentes (Cr 6+) son las especies metálicas más notorias que se sabe que se unen al ADN. Tanto el cromo trivalente como el hexavalente pueden causar mutaciones y el cromo hexavalente también es un carcinógeno conocido. Aunque las sales de Cr 6+ son solo ligeramente solubles, la reactividad del ion Cr 6+ es tan pronunciada que solo muy poca sal de cromo hexavalente ya es peligrosa.
La genotoxicidad del cromo trivalente se debe a la formación de entrecruzamientos entre proteínas y ADN. Cualquier molécula de ADN está rodeada de proteínas (histonas, proteínas reguladoras, cromatina). Cr 3+ se une a aminoácidos como cisteína, histidina y ácido glutámico por un lado, y a los grupos fosfato en el ADN por el otro, sin preferencia alguna por un nucleótido específico (base). El resultado es un enlace covalente entre el ADN y una proteína que inhibirá la transcripción o funciones reguladoras del segmento de ADN involucrado.
Otro metal que se sabe que interactúa con el ADN es el níquel. Aunque los efectos primarios del níquel son inducir reacciones alérgicas, también es un carcinógeno conocido. El mecanismo molecular exacto no es tan conocido como en el caso del cromo. El níquel podría reticular proteínas y ADN de la misma manera que el cromo, pero también se argumenta que la carcinogenicidad del níquel se debe al estrés oxidativo, lo que resulta en daño al ADN. Otro mecanismo sugerido es que el níquel podría interferir con el sistema de reparación del ADN.
Inhibición de bombas de iones
Muchos metales pueden competir con los metales esenciales durante la absorción o el transporte a través de las membranas. Un caso bien conocido es la competencia entre calcio y cadmio en la bomba de Ca 2+ ATPasa en la membrana basal de branquias de peces (Figura 2).
Las branquias de los peces sirven como blanco para muchos compuestos tóxicos acuíferos debido a su gran área de contacto con el agua, que consiste en varias membranas, cada una con infoldings para aumentar la superficie, y también su alta actividad metabólica que se deriva de sus importantes actividades reguladoras (captación de oxígeno, absorción de nutrientes y osmorregulación). El epitelio de una sola capa tiene dos tipos de células, una activa en la osmorregulación (llamadas células de cloruro) y otra activa en el transporte de nutrientes y oxígeno (llamadas células respiratorias). Existen fuertes uniones estrechas entre estas células para asegurar la impermeabilidad completa del epitelio a los iones. La membrana apical de las células respiratorias tiene muchas bombas y canales de captación (Figura 2). El calcio ingresa a las células a través de un canal de calcio (sin costos energéticos, siguiendo el gradiente). La concentración intracelular de calcio está regulada por una ATPasa de calcio en la membrana basal, que bombea calcio fuera de las células epiteliales hacia la sangre.
Figura en preparación
Figura 2. Representación esquemática de las células en un epitelio branquial de un pez, mostrando los flujos de calcio y cadmio. El calcio ingresa a la célula a través de canales de calcio en el lado apical, y es bombeado fuera de las células a la circulación por una ATPasa de calcio en la membrana basal. Los iones cadmio ingresan a las células también a través de los canales de calcio, pero inhiben la ATPasa basal de calcio, causando hipocalcemia en el resto del cuerpo. m = mucosa (lado apical), s = serosa (lado basal), BP = proteína de unión, mito = mitocondria, ER = retículo endoplásmico. De Verbost et al. (1989).
Los iones de cadmio transportados por el agua, que se asemejan a los iones calcio en su radio atómico, ingresan a la célula a través de los mismos canales apicales de calcio, pero posteriormente inhiben el transportador de calcio de la membrana basal por competencia directa con el calcio por el sitio de unión La consecuencia es una acumulación de calcio en las células respiratorias, y una falta de calcio en el cuerpo de los peces, lo que provoca una variedad de efectos secundarios; entre otros trastornos hormonales, mientras que una disminución severa del calcio plasmático es una causa directa de mortalidad. Este efecto del cadmio ocurre a concentraciones muy bajas (rango nanomolar), y explica la alta toxicidad de este metal para los peces. Mecanismos similares de hipocalcemia inducida por cadmio están presentes en las membranas branquiales de crustáceos y muy probablemente también en células epiteliales intestinales de muchas otras especies.
Interacción con constituyentes celulares esenciales
Existen diversos ligandos celulares fuera de proteínas o ADN que pueden unirse a metales. Entre estos se encuentran los ácidos orgánicos (malato, citrato), aminoácidos libres (histidina, cisteína) y glutatión. Los metales también pueden interferir con las funciones celulares de fosfato, hierro, calcio o zinc, por ejemplo reemplazando estos elementos de sus sitios de unión normales en enzimas u otras moléculas. Para ilustrar un caso de interacción con fosfato discutimos brevemente la toxicidad del arsénico. En sentido estricto, el arsénico no es un metal, ya que el óxido de arsénico puede participar tanto en reacciones formadoras de bases como de ácido. Junto con el antimonio y otros cuatro elementos menos conocidos, el arsénico se indica como un “metaloide”.
El arsénico es un potente tóxico; el trióxido de arsénico (As 2 O 3) es bien conocido por su alta toxicidad en mamíferos y su uso como rodenticida y conservante de la madera. También hay aplicaciones terapéuticas del trióxido de arsénico, contra ciertas leucemias y el arsénico suele estar implícito en los tratamientos homeopáticos. Los compuestos de arsénico se transportan fácilmente por todo el cuerpo, también a través de la barrera placentaria en mujeres embarazadas.
El arsénico puede presentarse en dos estados de valencia diferentes: arseniato (As 5+) y arsenito (As 3+). Los términos también se utilizan para indicar las oxisales, como arseniato férrico, FeASo 4, y arsenito férrico, FeASO 3. Dentro del cuerpo, el arsénico puede estar presente en estado oxidado así como reducido, dependiendo de las condiciones en la célula, y es convertido enzimáticamente a uno u otro estado por reductasas y oxidasas. También puede estar metilado por metiltransferasas. Las dos formas diferentes de arsénico tienen mecanismos de toxicidad bastante diferentes. El arseniato, AsO 4 3-, es un potente análogo del fosfato, mientras que el arsenito (AsO 3 3-) reacciona con los grupos SH en las proteínas, como los metales discutidos anteriormente. El arsenito también es un carcinógeno conocido; el mecanismo parece no depender de la unión al ADN, como en el caso del cromo, sino en la inducción del estrés oxidativo y la interferencia con la señalización celular.
La causa más común de intoxicación crónica por arsénico se debe a la inhibición de la enzima gliceraldehído fosfato deshidrogenasa (GAPDH). Esta es una enzima crítica de la glucólisis, convirtiendo el gliceraldehído-3-fosfato en 1,3-bifosfoglicerato. Sin embargo, en presencia de arseniato, GAPDH convierte el gliceraldehído-3-fosfato en 1-arseno-3-fosfoglicerato. En realidad el arseniato actúa como un análogo de fosfato para “engañar” a la enzima. El producto 1-arseno-3-fosfoglicerato no se involucra en la siguiente reacción glicolítica, que normalmente produce una molécula de ATP, pero vuelve a caer en arseniato y 3-fosfoglicerato, sin la producción de ATP, mientras que el arseniato liberado puede actuar nuevamente sobre la enzima de manera cíclica. El resultado es que la vía glicolítica está desacoplada de la producción de ATP. Huelga decir que esto significa una inhibición severa y a menudo fatal del metabolismo energético.
Patrones de especies de susceptibilidad a metales
Animales, plantas, hongos, protistas y procariotas difieren mucho en su susceptibilidad a los metales. Para dar algunos ejemplos:
- Se sabe que las lombrices y los caracoles son bastante sensibles al cobre; la ausencia de lombrices de tierra en huertos y viñedos donde se utilizan fungicidas que contienen cobre está bien documentada. Los caracoles no se pueden cultivar en agua que corre a través de forros que contienen cobre. Los hongos también son sensibles al cobre, lo que explica el uso del cobre en fungicidas. También muchas plantas son sensibles al cobre debido a los efectos sobre el crecimiento de las raíces. Entre los vertebrados, las ovejas son bastante sensibles al cobre, a diferencia de la mayoría de los otros mamíferos.
- Tanto los crustáceos como los peces son relativamente sensibles al zinc. Los mamíferos, sin embargo, apenas son sensibles al zinc.
- Los humanos son relativamente sensibles al plomo porque la alta exposición al plomo perturba el desarrollo del cerebro de los niños y se correlaciona con puntuaciones IQ bajas. Sin embargo, la mayoría de los invertebrados son bastante insensibles al plomo.
- Aunque muchos invertebrados son bastante sensibles al cadmio, la variación interespecie en la sensibilidad a este elemento es particularmente alta, incluso dentro del mismo linaje filogenético. El ácaro oribátido vivo del suelo Platynothrus peltifer es uno de los invertebrados más sensibles con respecto al efecto del cadmio en la reproducción, sin embargo Oppia nitens, también oribátido, es extremadamente tolerante al cadmio.
Al final, dichos patrones deben explicarse en términos de la presencia de dianas bioquímicas susceptibles, diferentes estrategias de almacenamiento y excreción, y diferentes mecanismos de defensa y secuestro. Sin embargo, por el momento no existe un marco general para comparar la variación de sensibilidad entre especies. Además, no hay relación entre acumulación y susceptibilidad; algunas especies que acumulan metales en gran medida (por ejemplo, cobre en isópodos) no son sensibles al mismo metal, mientras que otras, que no acumulan el metal, son bastante sensibles. La acumulación parece estar parcialmente relacionada con una estrategia de alimentación de especies (por ejemplo, las arañas absorben casi todos los alimentos (líquidos) que ingieren y cualquier metal en el alimento se acumulará en la glándula del intestino medio); la acumulación también está relacionada con requerimientos específicos de nutrientes (por ejemplo, cobre en isópodos, manganeso en algunos oribátidos ácaros). Finalmente, algunas poblaciones de algunas especies han evolucionado tolerancias específicas en respuesta a su vida en un ambiente contaminado con metales, además de las estrategias de acumulación y desintoxicación ya existentes.
Conclusión
Los metales no forman un grupo homogéneo. Su toxicidad implica reactividad hacia una gran variedad de dianas bioquímicas. A menudo, varios mecanismos actúan simultáneamente e interactúan entre sí. La inducción del estrés oxidativo es un denominador común, al igual que la reacción a grupos nucleofílicos en macromoléculas. La gran variedad de respuestas inducidas por metales los convierte en compuestos modelo interesantes para estudios toxicológicos.
Referencias
Cameron, K.S., Buchner, V., Tchounwou, P.B. (2011). Exploración de los mecanismos moleculares de genotoxicidad inducida por níquel y carcinogenicidad: una revisión de la literatura. Opiniones de Salud Ambiental 26, 81-92.
Ernst, W.H.O., Joosse-van Damme, E.N.G. (1983) Umwelbelastung durch Mineralstoffe. Fischer Verlag, Jena.
Singh, A.P., Goel, R.K., Kaur, T. (2011) Mecanismos relacionados con la toxicidad por arsénico. Toxicología Internacional 18, 87-93.
Verbost, P.M. (1989) Toxicidad del cadmio: interacción del cadmio con los mecanismos celulares de transporte de calcio Tesis de doctorado, Radboud Universiteit Nijmegen.
Mencionar tres clases diferentes de lesiones primarias por iones metálicos libres y causantes de toxicidad metálica. Incluir los metales que son más conocidos por causar cada tipo de lesión.
¿Es posible decidir a qué tipo de metal está expuesta una célula, en función del tipo de perturbación celular que se observa?
Varios invertebrados acumulan metales en un grado muy alto. Menciona algunos ejemplos y los metales que acumulan. ¿Estos animales también se encuentran entre los más sensibles a la toxicidad de los metales? Por favor explique.
“Los metales esenciales pueden ser regulados y por lo tanto son menos tóxicos que los metales xenobióticos” - Por favor comente esta tesis.
4.2.12. Tolerancia de metal
Autor: Nico M. van Straalen
Revisores: Henk Schat, Jaco Vangronsveld
Objetivos de aprendizaje
Deberías ser capaz de
- describir qué mecanismos de cambios en el tráfico de metales pueden contribuir a la tolerancia e hiperacumulación de metales
- explicar los factores moleculares asociados a la evolución de la tolerancia a metales en plantas y animales
- elaborar un dictamen sobre el tema del “rescate de la contaminación por evolución” en la evaluación de riesgos de metales pesados
Palabras clave: hiperacumulación, mecanismos de captación de metales, microevolución
Sinopsis
Algunas especies de plantas y animales han evolucionado poblaciones tolerantes a metales que pueden sobrevivir a exposiciones que son letales para otras poblaciones de la misma especie. La más conocida es la vegetación de metales pesados que crece en suelos metalíferos. El estudio de estos casos de “evolución en acción” ha revelado muchos aspectos del tráfico de metales en plantas, el transporte a través de membranas, las moléculas depuradoras de metales en la célula y la distribución subcelular de los metales, y cómo estos procesos han sido adaptados por selección natural para la tolerancia. Las variedades de plantas tolerantes a metales suelen depender de altas concentraciones de metal en el suelo y no crecen bien en suelos de referencia. Además, algunas especies de plantas muestran un grado extremo de acumulación de metal. En animales se ha demostrado tolerancia a los metales en algunos invertebrados que viven en estrecho contacto con suelos que contienen metales y esto generalmente se logra mediante la regulación alterada de proteínas depuradoras de metales como las metalotioneínas, o por duplicación de los genes correspondientes. Los estudios genómicos están ampliando nuestra perspectiva ya que la adaptación normalmente no se basa en un solo gen sino que incluye factores hipostáticos y modificadores.
Introducción
Como los metales no pueden ser degradados o metabolizados, la única manera de lidiar con el exceso potencialmente tóxico es almacenarlos o excretarlos. A menudo ambos mecanismos son operativos, siendo precedida la excreción por almacenamiento o recolección, pero los animales y las plantas difieren mucho en el énfasis en uno u otro mecanismo. Tanto los metales esenciales como los no esenciales están sujetos a todo tipo de mecanismos de tráfico con el objetivo de mantener extremadamente baja la concentración de iones libres biológicamente activos del metal. Aún así, apenas existe relación entre acumulación y tolerancia. Algunas especies tienen concentraciones tisulares bajas y son sensibles, otras tienen concentraciones tisulares bajas y son tolerantes, algunas acumulan metales y sufren las altas concentraciones, otras se acumulan y son extremadamente tolerantes.
Al igual que los mecanismos de biotransformación (ver la sección Variación Genética), los mecanismos de tráfico de metales muestran variación genética y dicha variación puede estar sujeta a evolución. Sin embargo, hay que señalar que sólo en un número limitado de plantas y especies animales han evolucionado poblaciones tolerantes a metales. Esto puede deberse a que la evolución de la tolerancia a los metales hace uso de mecanismos de tráfico de metales ya existentes, moderadamente eficientes, en las especies ancestrales. Esta interpretación es sugerida por la observación de que las variedades no tolerantes a metales de plantas tolerantes a metales ya tienen cierto grado de tolerancia al metal (mayor que las especies que nunca evolucionan variedades tolerantes a metales). Por lo que la distancia mutacional a la tolerancia al metal fue menor en los ancestros de las plantas tolerantes a metales que en las plantas “normales”.
La tolerancia real a los metales, donde la población tolerante a metales puede soportar órdenes de magnitud mayores que las poblaciones de referencia, y se ha vuelto dependiente de suelos ricos en metales, solo se encuentra en las plantas. La tolerancia al metal en los animales es de grado, más que de especie, y no viene con fenotipos reconocibles externamente. Lo más probable es que la combinación de una fuerte presión de selección, la imposibilidad de escapar por locomoción y la correcta variación genética preexistente expliquen por qué la tolerancia al metal en las plantas es mucho más prominente en comparación con los animales.
En esta sección discutiremos los diversos mecanismos que se ha demostrado que subyacen a la tolerancia al metal. La respuesta evolutiva a la exposición ambiental a metales es uno de los ejemplos clásicos de “evolución en acción”, junto a la resistencia a insecticidas en mosquitos y al melanismo industrial en mariposas.
Tolerancia al metal en plantas
Durante muchos años, muy probablemente ya desde que los humanos comenzaron a excavar minerales y utilizar metales para la fabricación de utensilios, cerámica y herramientas, se sabe que los suelos naturalmente ricos en metales albergan una vegetación específica tolerante a los metales. Esta “Schwermetallvegetation”, descrita en el libro clásico del botánico germano-holandés W.H.O. Ernst, consiste en una colección designada de especies vegetales, con representantes de diversas familias. Varias especies también tienen poblaciones sensibles a metales que viven en suelos normales, pero algunas, como la violeta de zinc europea, Viola calaminaria, están restringidas a suelos ricos en metales. Esto también se observa en las vegetaciones tolerantes a metales de Nueva Caledonia, Cuba, Zimbabue y Congo, que en gran medida consisten en especies endémicas tolerantes a metales (verdaderas metalófitas) que nunca se encuentran en suelos normales. Sin embargo, algunas especies comunes también desarrollaron ecotipos tolerantes a metales.
Las especies de plantas tolerantes a metales han ampliado su rango cuando los humanos comenzaron a excavar los minerales metálicos y ahora también se pueden encontrar extensamente en sitios mineros, bancos de arroyos enriquecidos con metales y alrededor de fundiciones de metal. Los suelos naturalmente enriquecidos con metales difieren de los suelos de referencia no solo en la concentración de metales sino también en otros aspectos, por ejemplo, calcio y humedad, por lo que la selección para la tolerancia al metal viene de la mano con la selección por varios otros factores.
La tolerancia al metal se restringe principalmente a las hierbas y hierbas, y (excepto algunas serpentinas tropicales) no se extiende a los árboles. Una vegetación metálica pesada es reconocible en el paisaje como una “pradera”, carente de árboles, con relativamente pocas especies de plantas y abundancia de metalófitas. En el pasado, se descubrieron minerales metálicos a partir de la presencia de tales metalófitas, una actividad llamada bioprospección.
Sabemos por bioquímica que diferentes metales están unidos a diferentes ligandos y siguen diferentes vías bioquímicas en tejidos biológicos (ver sección sobre acumulación de metales). Algunos metales (cadmio, cobre, mercurio) son “buscadores de azufre”, otros tienen afinidad por los ácidos orgánicos (zinc) y otros tienden a asociarse con tejidos ricos en calcio (plomo). Los metales esenciales como el cobre, el zinc y el hierro tienen sus propios mecanismos de transporte específicos para metales. A partir de estas observaciones se puede concluir que la tolerancia al metal también será específica del metal y que la tolerancia cruzada (tolerancia dirigida a un metal que causa tolerancia a otro metal como efecto secundario) es relativamente rara. Este es efectivamente el caso.
En muchos casos, las plantas tolerantes a metales no muestran las mismas características de crecimiento que las variedades no tolerantes de la misma especie. La pérdida de potencial de crecimiento a menudo se ha interpretado como un “costo de tolerancia”. Sin embargo, la investigación genética ha demostrado que el menor potencial de crecimiento de las metalófitas es una adaptación separada, para tratar los suelos metalíferos generalmente infértiles, y no existe un vínculo mecanicista con la tolerancia. No se han descrito costos metabólicos ni efectos pleiotrópicos negativos de la tolerancia a metales. El hecho de que las plantas tolerantes a metales no crecen bien en suelos limpios se explica por la regulación positiva constitutiva de los mecanismos de tráfico y compartimentación, provocando mayores requerimientos metálicos que no pueden cumplirse en suelos no metalíferos.
Otro dato llamativo es que las tolerancias metálicas en las mismas especies de plantas en diferentes sitios han evolucionado independientemente unas de otras. Las diversas poblaciones tolerantes a metales de una especie no descienden todas de una sola población ancestral, sino que son el resultado de una evolución local repetida. Que aún en diferentes poblaciones a veces los mismos loci se ven afectados por la selección natural, se atribuye a que, dados los antecedentes genéticos de la especie, solo hay un número limitado de vías para la tolerancia a los metales.
Un principio general final es que la tolerancia a los metales en las plantas a menudo se dirige hacia proteínas que transportan metales a través de membranas (membrana celular, tonoplasto). Los genes de dichos transportadores pueden estar duplicados, el equilibrio entre los transportadores de alta afinidad y las versiones de baja afinidad puede alterarse, su expresión puede estar regulada por incremento o regulación negativa, o las proteínas pueden dirigirse a diferentes compartimentos celulares.
Aunque aún faltan muchos detalles sobre los cambios genéticos responsables de la tolerancia en las plantas, el trabajo sobre la tolerancia al cobre en el campión vesical, Silene vulgaris, ilustra muchos de los puntos enumerados anteriormente. La planta tiene muchas poblaciones tolerantes a metales, de las cuales una encontrada en Imsbach, Alemania, muestra un grado extremo de tolerancia al cobre y también cierta tolerancia (evolucionada independientemente) al zinc y al cadmio. La zona es conocida por su “Bergbau” con actividades mineras históricas de cobre, plata y cobalto, pero también algunos depósitos de calamina más antiguos, lo que explica la tolerancia al zinc y al cadmio.
El trabajo genético de H. Schat y sus colegas ha demostrado que dos transportadores de cobre impulsados por ATP, designados HMA5I y HMA5II, están involucrados en la tolerancia al cobre de Silene. La proteína HMA5I reside en el tonoplasto para reubicar el cobre en la vacuola, mientras que HMA5II reside en el retículo endoplásmico. Cuando aparecen iones de cobre libres en la célula, el HMA5II se traslada del RE a la membrana celular y comienza a bombear cobre fuera de la célula. Durante el transporte de raíces a brotes (en los vasos de xilema) el cobre se une como un complejo de nicotianamina. Además, las metalotioneínas vegetales juegan un papel en la unión y el transporte del cobre en el floema y durante la redistribución de las hojas senescentes. La tolerancia al cobre en Silene ilustra el principio mencionado anteriormente de que la tolerancia a los metales se logra potenciando los mecanismos de transporte ya presentes, no mediante la evolución de nuevos genes.
Hiperacumulación de metales
Algunas plantas acumulan metales en grado extremo. Se conocen las metalófitas que crecen en suelos serpentinos, los cuales acumulan cantidades muy grandes de níquel. También se observa acumulación de cobre y cobalto en varias especies de plantas. Los hiperacumuladores no excluyen los metales sino que preferentemente los acumulan cuando la concentración en el suelo es extremadamente alta (> 50.000 mg de cobre por kg de suelo). La concentración de cobre de las hojas puede alcanzar valores superiores a 1000 μg/g. Un ejemplo muy extremo es una especie arbórea, Sebertia acuminata, que crece en la isla de Nueva Caledonia en suelo ultramafico con 0.85% de níquel, que produce un látex que contiene 11% de níquel en peso. Tales concentraciones extraordinarias imponen exigencias extremas sobre la eficiencia del tráfico de metales y así han atraído la atención de investigadores biológicos. En la vegetación de metales pesados de Europa occidental, los acumuladores de zinc están presentes en varias especies de los géneros Agrostis, Brassica, Thlaspi y Silene.
La mayor parte de la investigación experimental se realiza sobre las especies brasicaceas Noccaea (Thlaspi) caerulescens y Arabidopsis halleri, con Arabidopsis thaliana como modelo de referencia no acumulativo.
El transporte de metales en una planta implica una serie de etapas distintas, donde cada paso está regulado al alza en el hiperacumulador de metal. Esto se ilustra en la Figura 1 en Verbruggen et al. (2009) para la hiperacumulación de zinc en Thlaspi caerulescens.
- Absorción en células epiteliales radiculares; esto involucra transportadores de zinc ZIP4 e IRT1
- Transporte entre tejidos radiculares
- Carga del xilema radicular, mediante HMA4 y otros transportadores metálicos
- En el xilema, el zinc puede estar quelado por citrato, histidina de nicotianamina, o puede estar presente como iones libres
- Descarga del xilema en las hojas. Esto involucra proteínas YSL y otras.
- Transporte a vacuolas y quelación a quelantes específicos de vacuola como el malato, involucrando transportadores metálicos como HMA3, MTP1 y MHX.
Si bien se empiezan a conocer los componentes básicos del sistema, aún no está clara la cuestión de cómo se regula al alza toda la maquinaria de manera coherente.
Tolerancia al metal en animales
También en animales, se han reportado poblaciones tolerantes a metales de la misma especie, sin embargo, no existe una comunidad específica tolerante a metales con un conjunto designado de especies, como en plantas. Hay, sin embargo, acumuladores metálicos obvios entre los animales. Los más conocidos son los isópodos terrestres, que acumulan concentraciones muy altas de cobre en células designadas en sus hepatopáncreas, y algunas especies de ácaros oribátidos que acumulan cantidades muy altas de manganeso y zinc.
Uno de los factores investigados para explicar la tolerancia a metales en animales es una proteína de unión a metales, metalotioneína (MT). La duplicación génica de un gen MT ha sido implicada en la tolerancia de Daphnia y Drosophila al cobre. Además, la tolerancia a los metales puede deberse a una regulación transcripcional alterada. Este último mecanismo subyace en la evolución de la tolerancia al cadmio en el colgajo vivo del suelo, Orchesella cincta. El análisis genético detallado de este sistema modelo ha revelado que el promotor MT de O. cincta muestra un grado muy grande de polimorfismo, algunos alelos afectan los sitios de unión del factor de transcripción y provocan sobreexpresión de MT. El alelo promotor que confirió una fuerte sobreexpresión de MT tras la exposición al cadmio, tuvo una frecuencia significativamente mayor en poblaciones de O. cincta de suelos contaminados con metales (Figura 2).
Además de los colémbolos, también se ha descrito la evolución de la tolerancia a los metales para la lombriz, Lumbricus rubellus. En una población que vive en una zona minera desierta contaminada con plomo en Gales se distinguieron dos linajes sobre la base del gen COI y RFLP, Curiosamente, los dos linajes habían colonizado diferentes microhábitats de la zona, siendo uno de ellos incapaz de sobrevivir a altas concentraciones de plomo. Se observaron expresiones diferenciales para genes en el metabolismo de fosfato y calcio. Dos mutaciones cruciales en una proteína de transporte de calcio sugirieron que la tolerancia al plomo en L. rubellus se debe a la modificación del transporte de calcio, una diana lógica ya que a menudo se encuentra que el plomo y el calcio interactúan con el transporte del otro (ver la sección sobre acumulación de metal).
Conclusiones
El estudio de la tolerancia a los metales es un tema gratificante de la ecotoxicología evolutiva. Se han identificado varios mecanismos genéticos cruciales, pero en ninguno de los sistemas de estudio se dispone de una imagen completa de los mecanismos de tolerancia evolucionados. Cabe esperar que los estudios de todo el genoma puedan identificar la red completa responsable de la tolerancia, que probablemente incluya no solo genes principales, sino también factores hipostáticos y modificadores.
Referencias
Andre, J., King, R.A., Stürzenbaum, S.R., Kille, P., Hodson, M.E., Morgan, A.J. (2010). Diferenciación genética molecular en lombrices de tierra que habitan un paisaje heterogéneo contaminado con PB. Contaminación Ambiental 158, 883-890.
Ernst, W.H.O. (1974) Schwermetallvegetation der Erde. Gustav Fischer Verlag, Stuttgart.
Janssens, T.K.S., Roelofs, D., Van Straalen, N.M. (2009). Mecanismos moleculares de tolerancia a metales pesados y evolución en invertebrados. Ciencia de Insectos 16, 3-18.
Krämer, U. (2010). Hiperacumulación de metales en plantas. Revisión Anual de Biología Vegetal 61, 517-534.
Li, X., Iqbal, M., Zhang, Q., Espelta, C., Bliek, M., Hakvoort, H.W.J., Quatrocchio, F.M., Koes, R., Schat, H. (2017). Dos transportadores de cobre de Silene vulgaris que residen en diferentes compartimentos celulares confieren hipertolerancia al cobre por distintos mecanismos cuando se expresan en Arabidopsis thaliana. Nueva Fitólogo 215, 1102-1114.
Lopes, I., Baird, D.J., Ribeiro, R. (2005). Resistencia genéticamente determinada a niveles letales de cobre por Daphnia longispina: asociación con respuesta subletal y multiple/corresistencia. Toxicología y Química Ambiental 24, 1414-1419.
Van Straalen, N.M., Janssens, T.K.S., Roelofs, D. (2011). Microevolución de la tolerancia a tóxicos: de genes individuales al banco enredado del genoma. Ecotoxicología 20, 574-579.
Verbruggen, N., Herms, C., Schat, H. (2009) Mecanismos moleculares de la hiperacumulación de metales en plantas. Nueva Fitólogo 181, 759-776.
Describir la vía de los iones de zinc absorbidos por las plantas desde la solución del suelo hasta su destino final en la planta, y cómo se han modificado los diversos pasos en especies de plantas de hiperacumulación como Arabidopsis halleri.
Discutir la diferencia entre el cambio transregulatorio y el cambio cis-regulador en la evolución de la tolerancia a metales.
4.2.13. Vías de resultados adversos
Autor: Dick Roelofs
Revisores: Nico van Straalen, Dries Knapen
Objetivos de aprendizaje:
Deberías ser capaz de
- explicar el concepto de vía de resultados adversos.
- interpretar una representación gráfica de un AOP
- buscar en la base de datos AOPWiki eventos moleculares iniciadores, eventos clave y resultados adversos
Palabras clave: Evento de iniciación molecular, evento clave, ensayo in vitro, ensayo de alto rendimiento, vía
Introducción
En las últimas dos décadas la disponibilidad de datos moleculares, bioquímicos y genómicos ha aumentado exponencialmente. Actualmente se dispone de datos para una amplia gama filogenéticamente de organismos vivos, desde procariotas hasta humanos. Esto ha avanzado enormemente nuestro conocimiento y comprensión mecanicista de los sistemas biológicos, lo que es altamente beneficioso para diferentes campos de la investigación biológica como la genética, la biología evolutiva y las ciencias agrícolas. Al ser una ciencia biológica aplicada, la toxicología aún no ha aprovechado esta riqueza de datos, debido a que es difícil incorporar datos mecanicistas a la hora de evaluar la seguridad química en relación con la salud humana y el medio ambiente. Sin embargo, la sociedad está cada vez más preocupada por la liberación de químicos industriales con poca o ninguna información sobre peligros o riesgos. En consecuencia, es necesario considerar un número mucho mayor de productos químicos para los posibles efectos adversos en la salud humana y el funcionamiento de los ecosistemas. Para enfrentar este desafío es necesario desplegar enfoques rápidos, rentables y de alto rendimiento que puedan predecir la toxicidad potencial de las sustancias y reemplazar las pruebas tradicionales basadas en la supervivencia y reproducción que duran semanas o meses y a menudo requieren bastante mano de obra. Sin embargo, un desafío importante es vincular estos ensayos rápidos in vitro e in vivo con los criterios de valoración utilizados en la evaluación del riesgo actual. Este reto se recogió definiendo el marco de la vía de resultados adversos (AOP), propuesto por primera vez por Gerald Ankley y colaboradores de la Agencia de Protección Ambiental de Estados Unidos, US-EPA (Ankley et al., 2010).
El marco
El marco de AOP se define como una evolución de conceptos previos basados en caminos, especialmente mecanismos y modos de acción, para ensamblar y representar datos toxicológicos a través de niveles biológicos de organización (Ankley y Edwards, 2018). Un AOP es una representación gráfica de una serie de eventos clave medibles (KEs). Un evento clave es un cambio direccional medible en el estado de un proceso biológico. Los KEs pueden vincularse entre sí a través de relaciones de eventos clave (KER; ver Figura 1). El primer KE se representa como el "evento iniciador molecular" (MIE), y representa la interacción de la sustancia química con un receptor biológico que activa eventos clave posteriores. Lo ideal es que las relaciones de eventos clave se basen en evidencia causal. Una cascada de eventos clave puede eventualmente resultar en un resultado adverso (AO) a nivel individual o poblacional. El MIE y el AO son KEs especializados, pero tratados como cualquier otro KE en el marco de AOP.
El objetivo de una PAO es representar y describir, de manera simplificada, cómo las respuestas a nivel molecular y celular se traducen a impactos en el desarrollo, reproducción y supervivencia, los cuales son criterios de valoración relevantes en la evaluación del riesgo (Villeneuve et al., 2014). Se han definido cinco conceptos básicos en el desarrollo de AOP:
- Las AOP no son químicas específicas, son vías biológicas;
- Los AOP son modulares, se refieren a una cascada metabólica designada y definida, incluso si esa cascada interactúa con otros procesos biológicos;
- los AOP individuales se desarrollan como unidades pragmáticas;
- las redes de múltiples AOP que comparten KEs y KER son unidades funcionales de predicción para escenarios del mundo real; y
- Los AOP son documentos vivos que pueden cambiar con el tiempo basándose en nuevos conocimientos científicos.
Generalmente, los AOP son vías lineales simplificadas, pero se pueden organizar diferentes AOP en redes con nodos compartidos. Las redes de AOP son en realidad las unidades funcionales de predicción, ya que representan las complejas interacciones biológicas que ocurren en respuesta a la exposición a un tóxico o una mezcla de tóxicos. El análisis de las intersecciones (eventos clave compartidos) de diferentes AOP que conforman una red puede revelar conexiones biológicas inesperadas (Villeneuve et al., 2014).
Eventos de inicio molecular y eventos clave
Por lo general, un AOP consiste en solo un MIE y un AO, conectados entre sí por un número potencialmente ilimitado de KEs y KER. Se considera que el MIE es el primer anclaje de una AOP a nivel molecular, donde los estresores interactúan directamente con el receptor biológico. La identificación del MIE se basa principalmente en análisis químicos, análisis in silico o en datos químicos e in vitro. Por ejemplo, el MIE para los AOP relacionados con la activación del receptor de estrógenos implica la unión de sustancias químicas al receptor de estrógenos, desencadenando así una cascada de efectos en el metabolismo relacionado con las hormonas (ver la sección sobre Interrupción endocrina). El MIE para AOP relacionado con la sensibilización de la piel (ver más abajo) implica la interacción covalente de químicos con proteínas de la piel en las células de la piel, un evento llamado haptenización (Vinken, 2013).
Una amplia gama de datos biológicos puede apoyar la comprensión de los KEs. Por lo general, los KE tempranos (directamente ligados a los MIE) se evalúan mediante ensayos in vitro, pero pueden incluir datos in vivo a nivel celular, mientras que los KE intermedios y tardíos se basan en mediciones de tejidos, órganos o organismos completos (Figura 1). Las mediciones de eventos clave también están relacionadas con datos de cribado de alto rendimiento y/o datos generados por diferentes tecnologías ómicas. Aquí es en realidad donde entra el verdadero valor del marco AOP, ya que actualmente es el único marco capaz de alcanzar un nivel tan alto de integración de datos en el contexto de la evaluación de riesgos. Incluso es posible integrar datos comparativos de organismos filogenéticamente divergentes en mediciones de eventos clave, válidas entre especies, lo que podría facilitar la evaluación de la sensibilidad de las especies (Lalone et al., 2018). La producción final de AO suele estar representada por respuestas apicales, ya descritas como pautas estándar aceptadas e instrumentales en la toma de decisiones regulatorias, que incluyen criterios de valoración como el desarrollo, crecimiento, reproducción y supervivencia.
El desarrollo del marco AOP está actualmente respaldado por la Agencia de Protección Ambiental de Estados Unidos, los Centros Conjuntos de Investigación de la UE (ERC) y la Organización para la Cooperación y el Desarrollo Económicos (OCDE). Además, la OCDE ha patrocinado el desarrollo de una base de datos de búsqueda de acceso abierto AOPwiki (https://aopwiki.org/), que comprende más de 250 AOP con MIE, KE y KER asociados, y más de 400 factores estresantes. Los nuevos AOP se agregan regularmente. La base de datos también cuenta con un sistema para especificar la confianza a colocar en un AOP. Donde los KEs y los KER son apoyados por evidencias experimentales directas, específicamente diseñadas, se coloca en ellos una alta confianza. En otros casos, la confianza se considera moderada o baja, por ejemplo, cuando hay falta de datos de apoyo o evidencia contradictoria.
Ejemplo de caso: unión covalente a proteínas que conduce a sensibilización cutánea (AOP40)
La sensibilización cutánea se caracteriza por un proceso de dos etapas, una fase de sensibilización y una fase de provocación. El primer contacto de compuestos electrófilos con la piel modifica covalentemente las proteínas de la piel y genera una memoria inmunológica debido a las células T específicas de antígeno y alérgeno generadas. Durante la fase de provocación, el contacto repetido con el compuesto provoca la reacción alérgica definida como dermatitis de contacto alérgica, que generalmente se desarrolla en un efecto de por vida. Este es un punto final importante para la evaluación de la seguridad de los productos de cuidado personal, tradicionalmente evaluados mediante ensayos in vivo. A partir del cambio de opinión pública, la Agencia Química Europea (ECHA) decidió alejarse de las pruebas cutáneas de animales enteros y desarrolló estrategias de evaluación alternativas. Durante la sensibilización, el MIE tiene lugar cuando el químico ingresa a la piel, donde forma un complejo estable con proteínas portadoras específicas de la piel (complejos haptenos), que son inmunogénicas. Un KE posterior comprende la inflamación y la defensa oxidativa a través de una cascada de señalización llamada vía de señalización Keap1/Nrf2 (proteína asociada a ECH-similar a Kelch 1/factor nuclear eritroides 2 relacionado con factor 2). Al mismo tiempo, un segundo KE se define como activación y maduración de células dendríticas. Esto da como resultado el movimiento de las células dendríticas hacia los ganglios linfáticos, donde el complejo hapteno se presenta a las células T vívidas. El tercer KE describe la proliferación de células T específicas de hapteno y el posterior movimiento de células de memoria específicas de antígeno que circulan en el cuerpo. Tras un segundo contacto con el compuesto, estas células T de memoria secretan citocinas que causan una reacción inflamatoria que conduce a la AO incluyendo erupción roja, ampollas y ardor en la piel (Vinken et al., 2017). Esta AOP es designada AOP40 en la base de datos de vías de desenlace adverso.
Ahora se ha desarrollado un conjunto de ensayos in vitro de alto rendimiento para cuantificar los KEs intermedios en AOP40. Estos datos formaron la base para el desarrollo de un análisis de red bayesiana que puede predecir el potencial de sensibilización de la piel. Este ejemplo destaca el uso de datos derivados de vías organizados en un AOP, lo que finalmente conduce a un método alternativo de detección rápida que puede reemplazar a un método convencional usando experimentos con animales.
Referencias
Ankley, G.T., Bennett, R.S., Erickson, R.J., Hoff, D.J., Hornung, M.W., Johnson, R.D., Mount, D.R., Nichols, J.W., Russom, C.L., Schmieder, P.K., Serrrano, J.A., Tietge, J.E., Villeneuve, D.L. (2010). Vías de resultados adversos: Un marco conceptual para apoyar la investigación ecotoxicológica y la evaluación de riesgos. Toxicología y Química Ambiental 29, 730-741.
Ankley, G.T., Edwards, S.W. (2018). La vía de resultados adversos: un marco multifacético que apoya la toxicología del siglo XXI. Dictamen Actual en Toxicología 9, 1-7.
LaOne, C.A., Villeneuve, D.L., Doering, J.A., Blackwell, B.R., Transue, T.R., Simmons, C.W., Swintek, J., Degitz, S.J., Williams, A.J., Ankley, G.T. (2018). Evidencia de extrapolación cruzada de especies de resultados de ensayos de cribado de alto rendimiento a base de mamíferos. Ciencia y Tecnología Ambiental 18, 13960-13971.
Villeneuve, D.L., Crump, D., García-Reyero, N., Hecker, M., Hutchinson, T.H., LaOne, C.A., Landesmann, B, Lettieri, T., Munn, S., Nepelska, M., Ottinger, M.A., Vergauwen, L., Whelan, M. (2014). Desarrollo de la Vía Adversa de Utcome I: Estrategias y principios. Ciencias Toxicológicas 142, 312-320.
Vinken, M. (2013). El concepto de la vía de resultados adversos: Una herramienta pragmática en toxicología. Toxicología 312, 158-165.
Vinken, M., Knapen, D., Vergauwen, L., Hengstler, J.G., Angrish, M., Whelan, M. (2017). Vías de resultados adversos: una introducción concisa para toxicólogos. Archivos de Toxicología 91, 3697-3707.
¿Qué es un evento de iniciación molecular?
¿En qué parte de la cadena de eventos se alimentan los ensayos in vitro de alto rendimiento en los AOP?
¿Cuál es el objetivo final del concepto AOP?
¿Cuántos eventos clave intermedios se capturan en la AOP para la sensibilización de la piel?
¿Un AOP siempre se representa como una cadena lineal entre MIE y AO?
4.2.14. Variación genética en el metabolismo de los tóxicos
Autor: Nico M van Straalen
Revisores: Andrew Whitehead, Frank van Belleghem
Objetivos de aprendizaje:
Deberías ser capaz de
- explicar cuatro clases diferentes de variación y expresión del gen CYP, contribuyendo a las diferencias de especies
- explicar las asociaciones entre la actividad de biotransformación y ecologías específicas
- explicar cómo la variación genética en las enzimas de biotransformación puede conducir a la evolución de la tolerancia a los tóxicos
- describir la relevancia de los polimorfismos genéticos humanos para la medicina personalizada
Palabras clave: susceptibilidad a tóxicos; variación genética; evolución de biotransformación de la tolerancia a tóxicos
Conocimientos previos asumidos y módulos relacionados
- Biotransformación y procesamiento interno de productos químicos
- Mecanismos de defensa
- Erosión genética
Además, se necesitan conocimientos básicos de genética y biología evolutiva para entender este módulo.
Sinopsis
La susceptibilidad a los tóxicos a menudo muestra diferencias interindividuales asociadas con la variación genética. Si bien tales diferencias se consideran una molestia en las pruebas de toxicidad de laboratorio, son un aspecto inextricable de los efectos tóxicos en el ambiente. La variación puede deberse a polimorfismos en el sitio diana de acción tóxica, pero más a menudo las diferencias en las enzimas metabólicas y las tasas de excreción contribuyen a la variación interindividual. La estructura de los genes que codifican enzimas metabólicas, así como los polimorfismos en las regiones promotoras de dichos genes son fuentes comunes de variación genética. Bajo una fuerte presión de selección, las especies pueden evolucionar poblaciones tolerantes a tóxicos, por ejemplo, insectos a insecticidas y bacterias a antibióticos. En poblaciones humanas, los polimorfismos en las enzimas metabolizadoras de fármacos se mapean para proporcionar una base para terapias personales. Este módulo tiene como objetivo ilustrar algunos de los principios genéticos que explican la variación interindividual de la susceptibilidad tóxica y sus consecuencias evolutivas.
Introducción
Desde hace mucho tiempo se sabe que los sujetos humanos pueden diferir marcadamente en sus respuestas a los medicamentos: mientras que algunos pacientes apenas responden a una determinada dosis, otros reaccionan con vehemencia. Existen diferencias similares entre los sexos y entre grupos étnicos. Para evitar el fracaso del tratamiento por un lado y la sobredosis por el otro, tales diferencias personales han atraído el interés de los científicos farmacológicos. También la tendencia a desarrollar cáncer tras la exposición a sustancias químicas mutagénicas se debe en parte a la genética. Desde el auge de la ecología molecular en la década de 1990, los ecotoxicólogos han señalado que también existen diferencias interindividuales en las respuestas tóxicas en el medio ambiente.
Debido a la variación genética, la contaminación ambiental puede desencadenar un cambio evolutivo en la naturaleza. De la genética cuantitativa sabemos que cuando un rasgo se debe a muchos genes, cada uno con un efecto aditivo independiente sobre el valor del rasgo, la respuesta a la selección R, se relaciona linealmente con el diferencial de selección S según la fórmula: R = h 2 S, donde h 2 es una medida del heredabilidad del rasgo seleccionado (fracción de varianza genética aditiva relativa a la varianza fenotípica total). Dado que los tóxicos antropogénicos pueden actuar como agentes selectivos muy fuertes (S grande) se espera que siempre que h 2 > 0 haya adaptación. Sin embargo, la efectividad del “rescate evolutivo” de la contaminación se limita a aquellas especies que tienen la variación genética adecuada y la capacidad de aumentar rápidamente el tamaño de la población.
Polimorfismos de enzimas metabolizadoras de fármacos en humanos
Uno de los sistemas enzimáticos más importantes que contribuye al metabolismo de los químicos xenobióticos es la familia del citocromo P450, una clase de proteínas localizadas en el retículo endoplásmico liso de la célula y que actúan en cooperación con varias otras proteínas. El citocromo P450 oxidará el sustrato y potenciará su solubilidad en agua (llamada reacción de fase I), y en muchos casos lo activará para reacciones posteriores que implican conjugación con un compuesto endógeno (reacciones de fase II). Estos procesos generalmente conducen a la desintoxicación y aumento de la excreción de sustancias tóxicas. La bioquímica del metabolismo farmacológico se discute en detalle en la sección sobre Metabolismo xenobiótico y defensa.
El genoma humano tiene 57 genes que codifican una proteína P450. Los genes son comúnmente designados como “CYP”. Otros organismos, especialmente insectos y plantas tienen muchos más CYP. Por ejemplo, el genoma de Drosophila codifica 83 genes P450 funcionales y el genoma de la planta modelo Arabidopsis tiene 244 CYP. Con base en la similitud de secuencias, los CYP se clasifican en 18 familias y 43 subfamilias, pero aún no hay acuerdo sobre la posición de varios genes CYP en invertebrados inferiores. La complejidad se ve potenciada por duplicaciones específicas de ciertos linajes evolutivos, creando un patrón complicado de ortólogos (homólogos por descendencia de un ancestro común) y parálogos (homólogos por duplicación en el mismo genoma). Además de las enzimas funcionales también es común encontrar muchos pseudogenes CYP en un genoma. Los pseudogenes son secuencias de ADN que se asemejan a genes funcionales, pero están mutados y no dan como resultado proteínas funcionales).
La expresión de las enzimas CYP es marcadamente específica de tejido. A menudo, la expresión de CYP es alta en tejidos epiteliales (pulmón, intestino) y órganos con actividad metabólica designada (hígado, riñón). En el cuerpo humano, el hígado es el principal órgano metabólico y es conocido por su extensa expresión de CYP. Las enzimas P450 también difieren en su inducibilidad por clases de químicos y en su especificidad de sustrato.
A menudo se asume que la versatilidad de los genes CYP de un organismo es un reflejo de su ecología. Por ejemplo, los insectos herbívoros que consumen plantas de diferentes tipos con muchos repelentes alimentarios diferentes deben aprovechar una amplia diversidad de genes CYP. También se ha demostrado que la actividad de las enzimas CYP entre los organismos terrestres es, en general, mayor que entre los organismos acuáticos y que las aves alimentadoras de plantas tienen mayores actividades de biotransformación que las aves depredadoras.
Uno de los genes CYP mejor investigados, especialmente debido a su fuerte inducibilidad y participación en el metabolismo xenobiótico, es el CYP1A1 de mamíferos. En humanos, la inducción de este gen se asocia con un mayor riesgo de cáncer de pulmón por fumar, y con otros cánceres, como el cáncer de mama y el cáncer de próstata. El CYP1A1 humano se localiza en el cromosoma 15 y codifica 251 aminoácidos en siete exones (ver Figura 2 en Zhou et al., 2009). Para este gen se han descrito 133 polimorfismos de un solo nucleótido (SNP, variaciones en un solo nucleótido que ocurren en una posición específica del genoma), de los cuales 23 son no sinónimos (provocando una sustitución de un aminoácido en la proteína).
Muchos de estos SNP tienen relevancia médica. Por ejemplo, un SNP bastante común en el exón 7 cambia el codón 462 de isoleucina a valina. El alelo sustituido se llama CYP1A1*2A, y esto ocurre con una frecuencia del 19% en la parte caucásica de la población humana. La variante alélica de la enzima tiene una mayor actividad hacia el 17β-estradiol y es un factor de riesgo para varios tipos de cáncer. Sin embargo, la expresión de tales rasgos puede variar de una población a otra, y también puede interactuar con otros factores de riesgo. Por ejemplo, el CYP1A1*2A es un factor de riesgo de cáncer de cuello uterino en mujeres con antecedentes de tabaquismo en la población polaca, pero el mismo SNP puede no ser un factor de riesgo en otra población o entre personas con un estilo de vida para no fumadores. En genética estos efectos se conocen como epistasis: el efecto fenotípico de la variación genética en un locus depende del genotipo de otro locus. Este es también un ejemplo de interacción genotipo por ambiente, donde el efecto fenotípico de una variante genética depende del ambiente (hábito de fumar). En toxicología se sabe que los polimorfismos de las enzimas de biotransformación de fase II pueden contribuir significativamente a la interacción epistática con genes CYP. Desentrañar todas estas interacciones complicadas es un campo de investigación muy activo en genética médica humana.
Variación del citocromo P450 entre especies
La comparación de genes CYP en diferentes especies ha revelado una evolución enormemente rápida de esta familia de genes, con muchas duplicaciones específicas de linaje. Esto indica fuertes presiones selectivas impuestas por la necesidad de desintoxicar las sustancias ingeridas con la dieta. Especialmente los animales herbívoros están constantemente expuestos a tales compuestos, sintetizados por las plantas para disuadir la alimentación. También vemos profundos cambios en los genes CYP asociados con transiciones evolutivas como la colonización de hábitats terrestres por los diversos linajes de artrópodos. Dicha variación natural, inducida por las toxinas vegetales y los requerimientos del hábitat, también es relevante en las respuestas a los tóxicos.
En general, la variación de las enzimas de biotransformación se puede clasificar en cuatro categorías principales
- Variación en la estructura de los genes, por ejemplo sustituciones que alteran la afinidad de unión a sustratos; dicha variación discrimina los diversos genes CYP.
- Variación del número de copias; la duplicación suele conducir a un aumento de la capacidad enzimática; este proceso ha sido enormemente importante en la evolución del CYP. Debido a que las duplicaciones de genes CYP suelen ser específicas del linaje evolutivo, surge un patrón complicado de parálogos (duplicados dentro del mismo genoma) y ortólogos (genes comunes por descendencia, compartidos con otras especies).
- Variación del promotor, por ejemplo, debido a la inserción de transposones o cambios en el número o disposición de los sitios de unión al factor de transcripción. Esto cambia la cantidad de proteína producida a partir de una copia génica por regulación transcripcional alterada.
- Variación en la estructura, acción o activación de reguladores transcripcionales. La transcripción de las enzimas de biotransformación suele ser inducida por una vía de señalización activada por el compuesto a metabolizar (ver la sección Metabolismo y defensa xenobióticos), y esta vía puede mostrar variación genética.
Para ilustrar la complicada evolución de los genes de biotransformación, discutimos brevemente los CYP del cormorán común, Phalacrocorax carbo. Se trata de un ave conocida por su dieta estrecha (peces) y su extraordinario potencial de acumulación de compuestos relacionados con las dioxinas (PCB, PCDD y PCDF). Los toxicólogos ambientales han identificado dos genes CYP1A en el cormorán, llamados CYP1A4 y CYP1A5. Resulta que CYP1A4 es homólogo por descenso (ortólogo) al CYP1A1 de mamífero mientras que CYP1A5 es un ortólogo del CYP1A2 de mamífero. Sin embargo, las ortologías no se revelan mediante análisis filogenéticos comunes si se usa toda la secuencia codificante en el alineamiento (ver Figura 3 en Kubota et al., 2006). Esto es consecuencia de un proceso llamado conversión de genes interparalog, que tiende a homogeneizar secuencias de ADN de copias génicas localizadas en el mismo cromosoma. Esto disminuye la variación de secuencia entre los parálogos, y crea estructuras génicas quiméricas, que son más similares entre sí de lo esperado por sus relaciones filogenéticas. Si se elabora un árbol filogenético utilizando una sección del gen que permaneció fuera de la conversión génica, se revelan las verdaderas relaciones filogenéticas (ver Figura 3 en Kubota et al., 2006).
Resistencias mediadas por citocromo P450
Los polimorfismos del citocromo P450 también están implicados en ciertos tipos de resistencia a insecticidas. Hay muchas maneras en que los insectos y otros artrópodos pueden volverse resistentes e incluso pueden estar presentes varios mecanismos en una misma cepa resistente. La alteración del sitio diana (haciendo que la diana sea menos susceptible al insecticida, por ejemplo, acetilcolinesterasa alterada, sustituciones en el receptor GABA, etc.) parece ser el mecanismo más probable de resistencia, sin embargo, tales cambios a menudo vienen con costos sustanciales ya que pueden disminuir la función natural del target (en genética esto se llama pleiotropía). El aumento del metabolismo no suele aportar costos metabólicos y aquí es donde entran en juego los citocromos P450. Un sistema modelo para investigar la genética de tales mecanismos es la resistencia al DDT en la mosca de la fruta, Drosophila melanogaster.
En una cepa de Drosophila resistente a DDT, todos los genes CYP se seleccionaron para una mayor expresión y se demostró que la resistencia a DDT se debió a una variante altamente regulada por incremento de un solo gen, CYP6G1. El análisis posterior mostró que el promotor del gen llevaba una inserción con fuerte similitud con un elemento transponible de la familia Accord. La inserción de este elemento provoca una sobreexpresión significativa y una alta tasa de síntesis de proteínas que permite a la mosca degradar rápidamente una dosis de DDT. El hecho de que un simple cambio, en un solo alelo, pueda subyacer un fenotipo tan distintivo como la resistencia a los pesticidas es una lección notable para la toxicología molecular.
Un estudio reciente sobre el killifish, Fundulus heteroclitus, a lo largo de la costa este de Estados Unidos ha revelado un patrón de resistencia mucho más complicado. Las poblaciones de estos peces viven en estuarios, algunos con sedimentos severamente contaminados, que contienen altas concentraciones de bifenilos policlorados (PCB) e hidrocarburos aromáticos policíclicos (HAP). Los peces killifish de los ambientes contaminados son mucho más resistentes a la toxicidad de los compuestos modelo PCB126 y benzo (a) pireno. Esta resistencia está relacionada con mutaciones en el gen que codifica el receptor de hidrocarburos arílicos (AHR), la proteína que se une a los HAP y ciertos metabolitos de PCB y activa la expresión de CYP. También las mutaciones en una proteína llamada proteína que interacciona con el receptor de aril-hidrocarburo (AIP), una proteína que se combina con AHR para asegurar la unión del ligando, contribuyen a la regulación a la baja de la vía CY1A1. El resultado neto es que el killifish CYP1A1 muestra solo una inducción moderada por PCB y HAP y se evitan los efectos dañinos de los metabolitos reactivos. Sin embargo, dado que la caída directa de CYP1A1 no proporciona resistencia, todavía no está claro si los efectos beneficiosos de las mutaciones en AHR realmente actúan a través de un efecto sobre CYP1A1.
Curiosamente, las diversas poblaciones de killifish muestran al menos tres deleciones diferentes en los genes AHR (Figura 1). Además, las poblaciones tolerantes muestran diversos grados de duplicación del CYP1A1; en una población incluso hay ocho parálogos presentes. Esto puede interpretarse como adaptaciones compensatorias que aseguran un nivel constitutivo basal de la proteína CYP1A1 para realizar actividades metabólicas rutinarias. El ejemplo de killifish muestra un maravilloso caso de interacción entre retoques genéticos y una fuerte selección que emana de un ambiente contaminado.
Conclusión
En este módulo nos hemos centrado en la variación genética en la enzima fase I, el citocromo P450. Una complejidad similar se encuentra detrás de las enzimas de fase II y los diversos transportadores inducidos por xenobióticos (fase III). Aún así, los ejemplos de P450 bastan para demostrar que la maquinaria del metabolismo xenobiótico muestra un grado muy grande de variación genética, así como diferencias de especies por duplicaciones, deleciones, conversión génica y selección específica de linaje. La variación reside tanto en la variación del número de copias, alteración de secuencias codificantes como en secuencias promotoras o potenciadoras que afectan la expresión de las enzimas. Dicha variación genética es la plantilla para la evolución. En ambientes contaminados, a veces se selecciona la expresión mejorada para (para neutralizar compuestos tóxicos), pero a veces también se selecciona la expresión atenuada (para evitar la producción de intermedios tóxicos). En el genoma humano, muchos de los polimorfismos tienen un significado médico, determinando un perfil personal del metabolismo de los fármacos y tendencias a desarrollar cáncer.
Referencias
Bell, G. (2012). El rescate evolutivo y los límites de la adaptación. Transacciones filosóficas de la Real Sociedad B 368, 2012.0080.
Daborn, P.J., Yen, J.L., Bogwitz, M.R., Le Goff, G., Feil, E., Jeffers, S., Tijet, N., Perry, T., Heckel, D., Batterham, P., Feyereisen, R., Wilson, T.G., Ffrench-Constant, R.H. (2002). Un solo alelo P450 asociado a resistencia a insecticidas en Drosophila. Ciencia 297, 2253-2256.
Feyereisen, R. (1999). Enzimas del insecto P450. Revisión Anual de Entomología 44, 507-533.
Goldstone, H.M.H., Steeman, J.J. (2006). Una historia evolutiva revisada de la subfamilia CYP1A: duplicación génica, conversión génica y selección positiva. Revista de Evolución Molecular 62, 708-717.
Kubota, A., Iwata, H., Goldstone, H.M.H., Kim, E.-Y., Steeman, J.J., Tanabe, S. (2006). Citocromo P450 1A1 y 1A5 en cormorán común (Phalacrocorax carbo): relaciones evolutivas e implicaciones funcionales asociadas con dioxinas y compuestos relacionados. Ciencias Toxicológicas 92, 394-408.
Reid, N.M., Proestou, D.A., Clark, B.W., Warren, W.C., Colbourne, J.K., Shaw, J.R., Hahn, M., Nacci, D., Oleksiak, M.F., Crawford, D.L., Whitehead, A. (2016). El panorama genómico de rápida adaptación evolutiva repetida a la contaminación tóxica en peces silvestres Science 354, 1305-1308.
Preissner, S.C., Hoffmann, M.F., Preissner, R., Dunkel, R., Gewiess, A., Preissner, S. (2013). Enyzmes polimórficos del citocromo P450 (CYP) y su papel en la terapia personalizada. PLoS ONE 8, e82562.
Roszak, A., Lianeri, M., Sowinska, A., Jagodzinski, P.P. (2014). CYP1A1 Polimorfismo Ile462Val como factor de riesgo en el desarrollo del cáncer cervicouterino en las poblaciones polacas. Diagnóstico y Terapia Molecular 18, 445-450.
Taylor, M., Feyereisen, R. (1996). Biología molecular y evolución de la resistencia a tóxicos. Biología Molecular y Evolución 13, 719-734.
Walker, C.H., Ronis, M.J. (1989). Las monooxigenasas de aves, reptiles y anfibios. Xenobiotica 19, 1111-1121.
Zhou, S.-F., Liu J.-P., Chowbay, B. (2009). Polimorfismo de enzimas del citocromo P450 humano y su impacto clínico. Metabolismo de los Medicamentos Reseñas 41, 89-295.
Comentario en: “En el gen 1A1 del citocromo P450 humano se han descrito 133 polimorfismos de un solo nucleótido, de los cuales 23 son no sinónimos”.
- ¿Qué es un “polimorfismo de un solo nucleótido” (SNP)?
- ¿Cuál es la diferencia entre un SNP sinónimo y un SNP no sinónimo?
- ¿Cuáles podrían ser las diferencias de sustituciones de nucleótidos (1) en el promotor de CYP1A1, (2) en uno de los intrones del gen CYP1A1, (3) en uno de los exones de CYP1A1?
- ¿Un solo cambio de aminoácido en la proteína del citocromo P450 tiene alguna relevancia médica?
Algunas poblaciones de killifish a lo largo de la costa atlántica de Estados Unidos muestran una resistencia muy alta contra contaminantes orgánicos como los PCB similares a las dioxinas. La investigación genética ha demostrado que estas poblaciones resistentes tienen una deleción en el gen que codifica el receptor de hidrocarburos arílicos (Ahr). Explique por qué tal mutación puede causar resistencia a los PCB similares a las dioxinas.
Comentar el concepto de “Rescate evolutivo de la contaminación” - la evolución de la tolerancia como una forma de disminuir los efectos tóxicos de la contaminación.