16.1: Teoría de la espectrometría de absorción infrarroja
- Page ID
- 78696

Comprensión del espectro IR
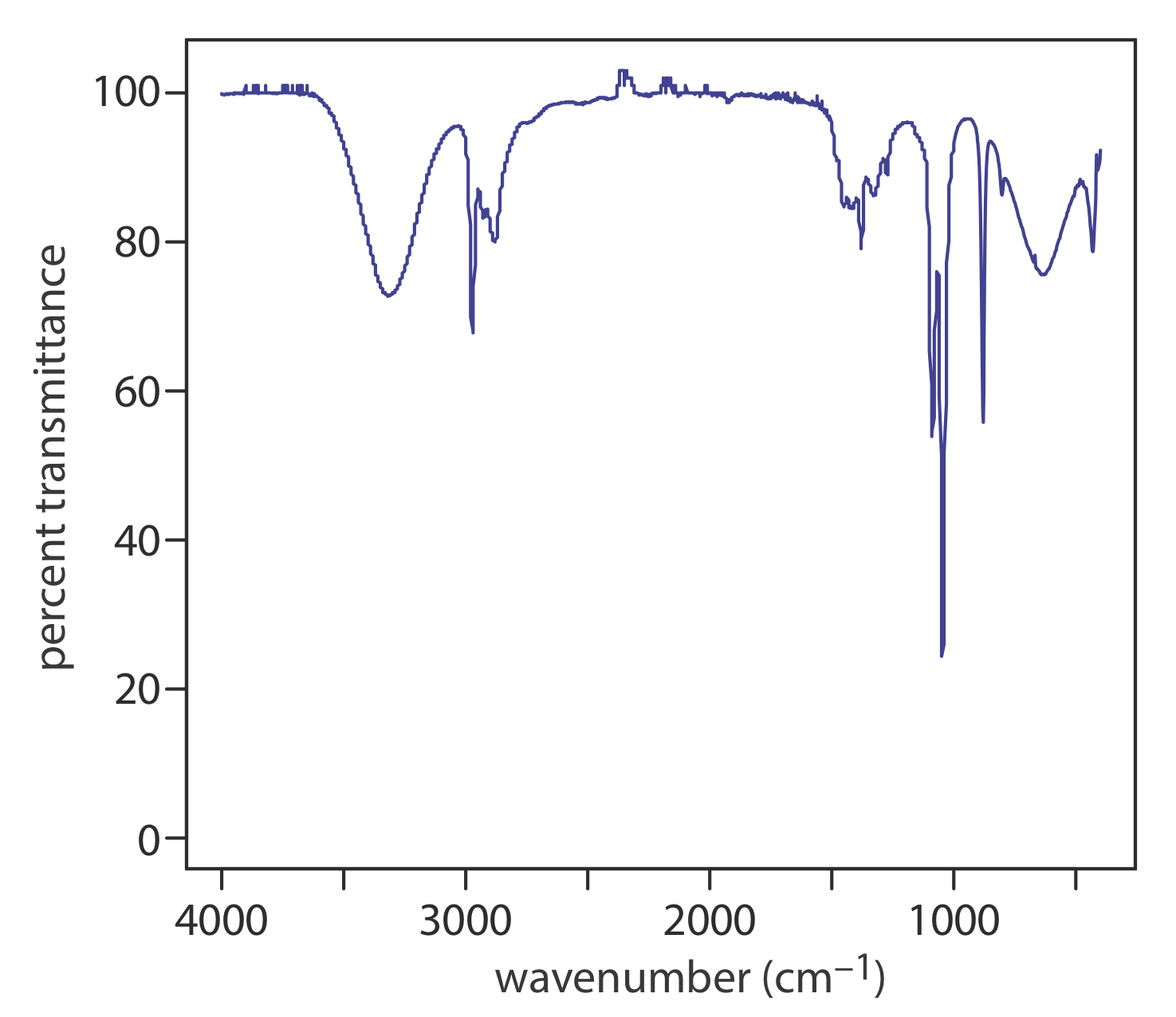
La figura\(\PageIndex{1}\) muestra el espectro infrarrojo para etanol. A diferencia de un espectro de absorbancia UV/Vis, el eje y se muestra como porcentaje de transmitancia (%T) en lugar de absorbancia, reflejando el hecho de que IR se usa más con fines cualitativos que cuantitativos, donde la ley de Beer, que es una función lineal de concentración (\(A = \epsilon b C\)) hace absorbancia la medida más útil. El eje x para un espectro IR generalmente se da en números de onda,\(\overline{\nu} = \lambda^{-1}\), con unidades de cm —1. Los picos en un espectro IR se invierten en relación con el espectro de absorbancia; es decir, descienden de una línea base de 100% T en lugar de elevarse desde una línea base de absorbancia cero.
Cambios de dipolo
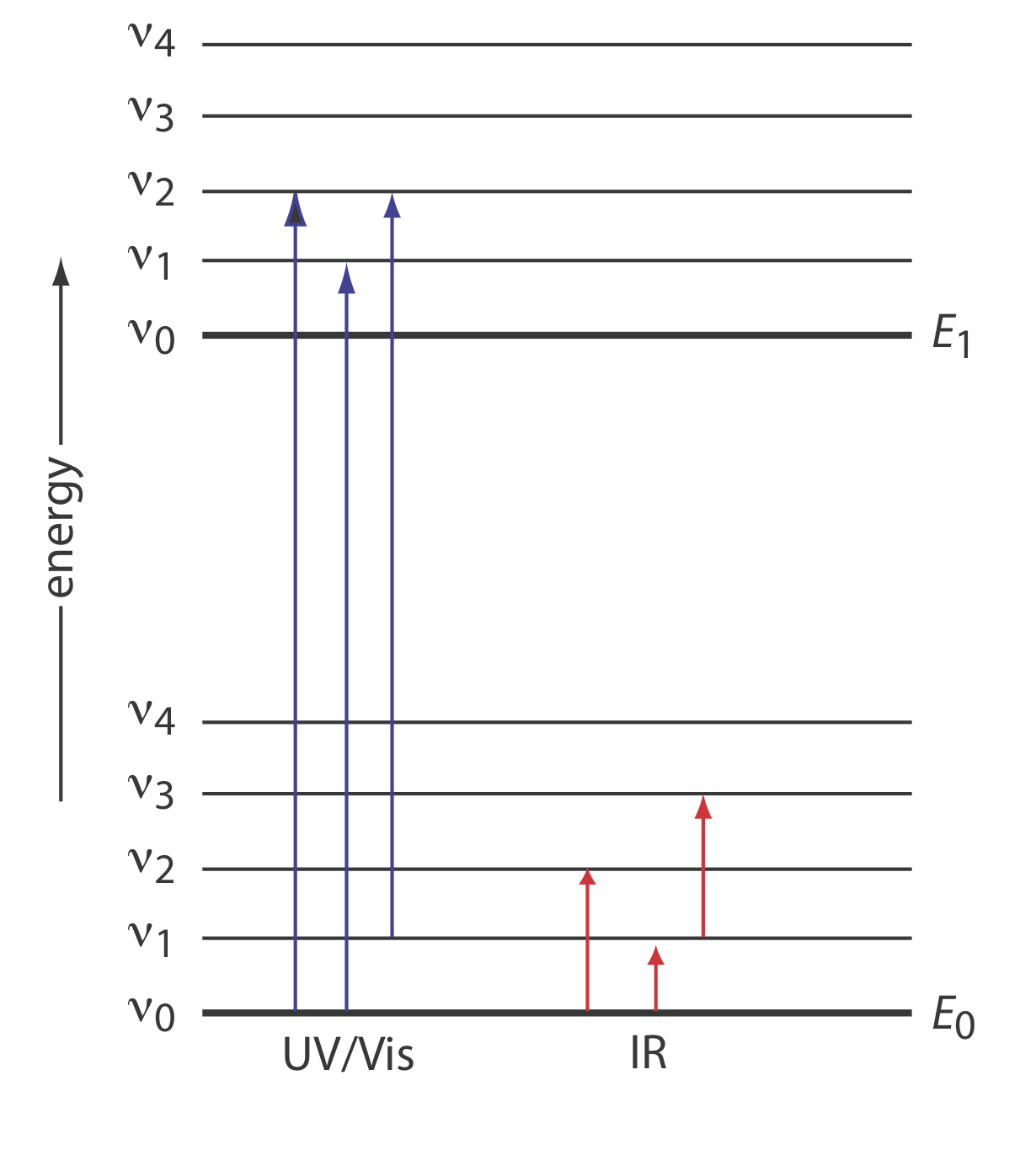
La energía de un fotón de radiación infrarroja (ver Figura\(\PageIndex{2}\)) no es suficiente para afectar un cambio en los niveles de energía electrónica de los electrones, como en las espectroscopias UV/Vis de absorción atómica o molecular o de emisión cubiertas en los Capítulos 9, 10 y 12—15. En cambio, la radiación infrarroja se limita a cambios en los estados de energía vibratoria de moléculas e iones moleculares. Para absorber un fotón IR, la especie absorbente debe experimentar un cambio en su momento dipolar, lo que permite que la oscilación en el campo eléctrico del fotón interactúe con una oscilación en carga dentro de la especie absorbente. Si las dos oscilaciones tienen la misma frecuencia, entonces la absorción es posible.
Cada estado de energía vibracional en la Figura\(\PageIndex{2}\) también tiene un conjunto de estados de energía rotacional, lo que significa que el pico para un cambio particular en los niveles de energía vibratoria puede consistir en una serie de líneas estrechamente espaciadas, una para cada uno de varios cambios en la energía rotacional. Debido a que la rotación es difícil para analitos que en formas líquidas o sólidas, solemos ver una sola línea de absorción amplia; por esta razón, consideraremos solo las transiciones vibracionales en este capítulo.
Tipos de vibraciones moleculares
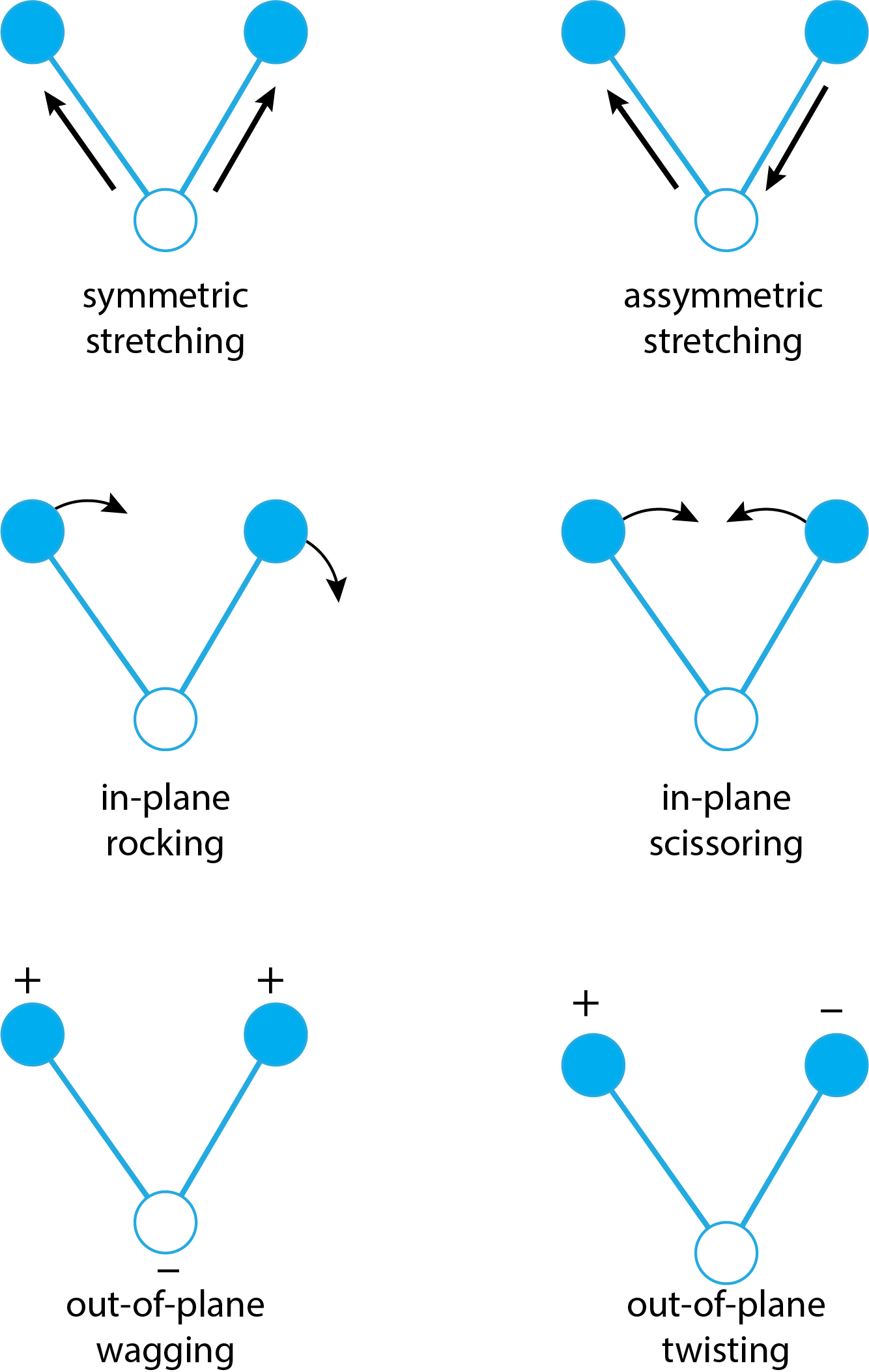
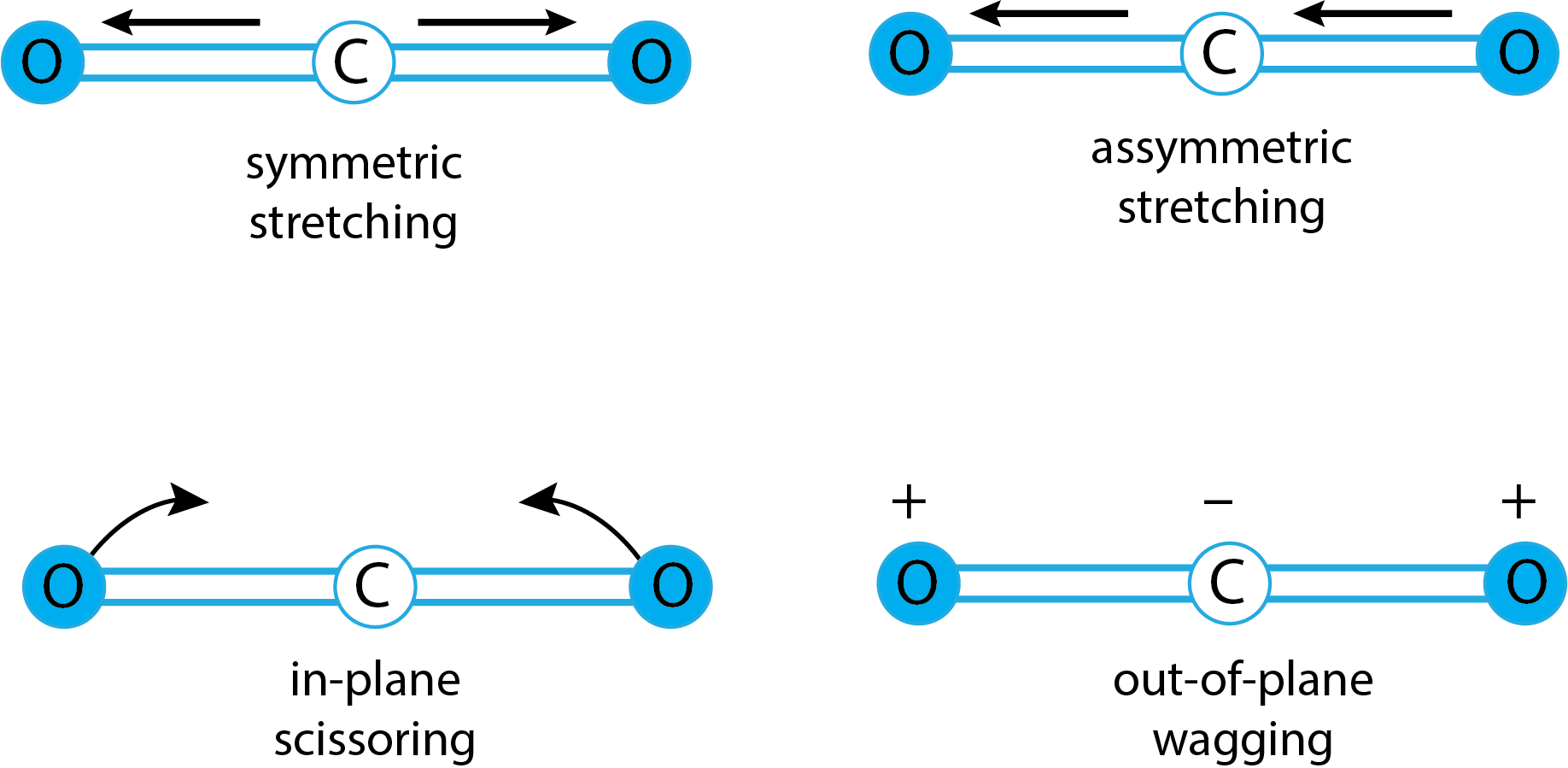
Aunque tendemos a pensar que los átomos de una molécula están rígidamente fijados en el espacio entre sí, los átomos individuales están en un estado de movimiento constante: las longitudes de enlace aumentan y disminuyen al estirarse y comprimirse, y los ángulos de enlace cambian como resultado de la flexión de los enlaces en relación con cada uno otro. La figura\(\PageIndex{3}\) muestra dos tipos diferentes de estiramiento (simétrico y asimétrico) y cuatro tipos diferentes de flexión (balanceo en plano, tijera en plano, movimiento fuera del plano y torsión fuera del plano).
Incluso una molécula simple puede tener muchos modos vibracionales que dan lugar a un pico en el espectro IR, como es el caso del etanol (Figura\(\PageIndex{1}\)). El número de modos vibracionales normales posibles para una molécula lineal es\(3N - 5\), donde N es el número de átomos, y\(3N - 6\) para una molécula no lineal. El etanol, por ejemplo, tiene\(3 \times 9 - 6 = 21\) posibles modos vibracionales. Como veremos más adelante en esta sección, algunos de estos modos pueden no conducir a un cambio en el momento dipolar, disminuyendo el número de picos en el espectro IR.
¿Por qué una molécula no lineal tiene modos\(3N - 6\) vibracionales? Considerar una molécula de metano, CH 4. Cada uno de los cinco átomos del metano puede moverse en una de tres direcciones (x, y y z) para un total de\(3 \times 5 = 15\) diferentes formas en las que los átomos de la molécula pueden moverse. Una molécula puede moverse de tres maneras: puede moverse de un lugar a otro, lo que llamamos movimiento traslacional; puede girar alrededor de un eje, al que llamamos movimiento rotacional; y sus enlaces pueden estirarse y doblarse, lo que llamamos movimiento vibracional. Debido a que toda la molécula puede moverse en las direcciones x, y y z, tres de las 15 formas diferentes de movimiento del metano son traslacionales. Además, la molécula puede rotar alrededor de sus ejes x, y y z, lo que representa tres formas adicionales de movimiento. Esto deja modos\(15 - 3 - 3 = 9\) vibracionales. Una molécula lineal, como el CO 2, tiene modos\(3N - 5\) vibracionales porque puede girar alrededor de solo dos ejes.
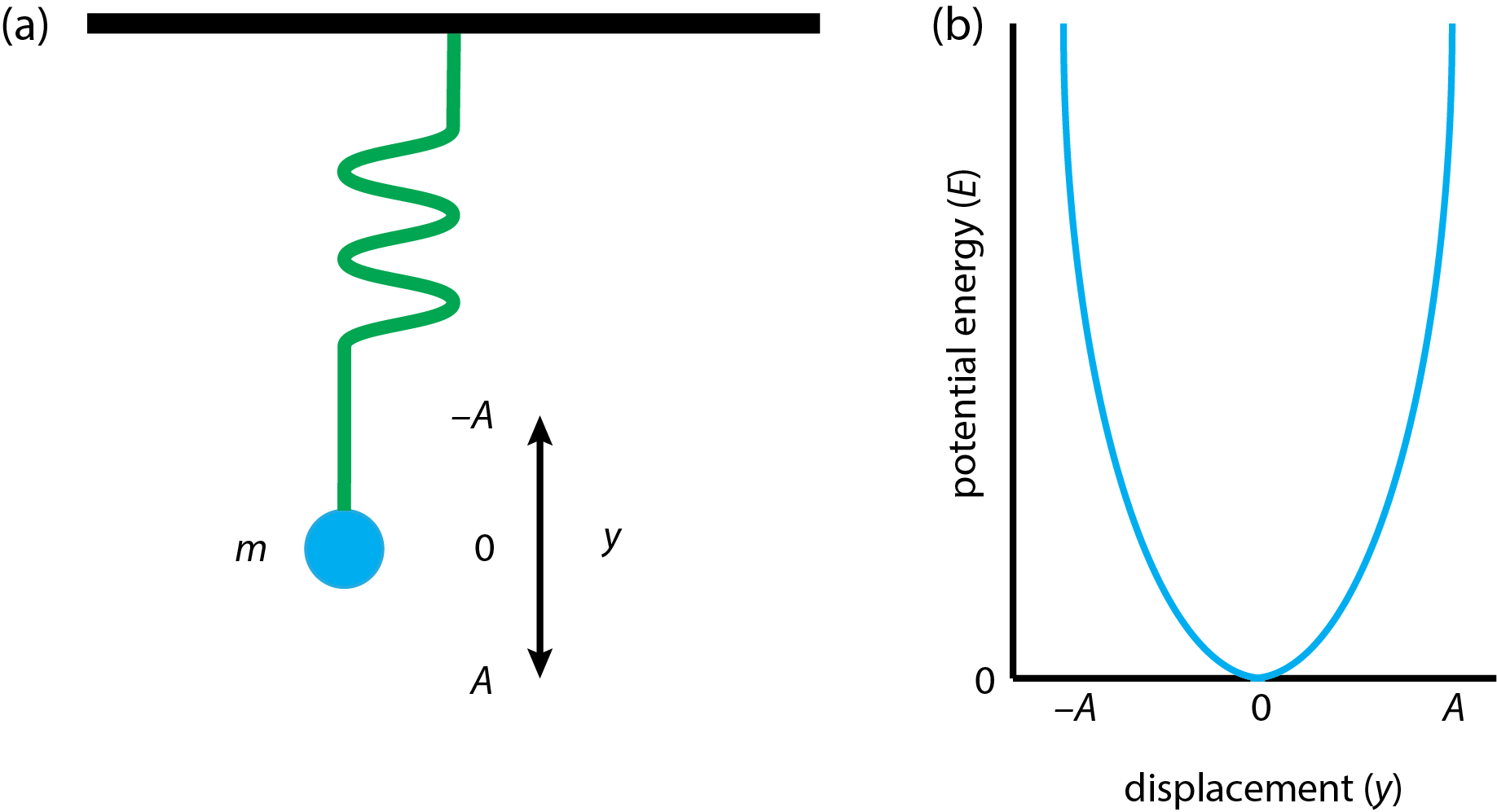
Modelo mecánico de estiramiento en una molécula diatómica
El sistema modelo más simple para el estiramiento y compresión de una unión es un peso con una masa, m, unido a un resorte ideal que cuelga del techo como se muestra en la Figura\(\PageIndex{4}a\). Si tiramos de la masa y luego la liberamos, iniciamos un simple movimiento armónico oscilante que podemos modelar usando la ley de Hooke. Si desplazamos el peso por una distancia, y, entonces la fuerza, F, que actúa sobre el peso es
\[F = - k y \label{hookeslaw} \]
donde\(k\) está la constante de fuerza del resorte, una medida de la elasticidad del resorte. El signo negativo en la Ecuación\ ref {hookeslaw} indica que esta es la fuerza necesaria para restaurar el resorte a su posición original; es decir, la fuerza está en la dirección opuesta a nuestra acción de tirar hacia abajo sobre el peso.
Energía potencial de un oscilador armónico
Tomemos la energía potencial, E, de la primavera y el peso como 0 cuando están en reposo (y = 0). Si tiramos hacia abajo en el peso por una distancia de\(dy\), entonces el cambio en la energía potencial del sistema,\(dE\), debe aumentar por el producto de la fuerza y la distancia
\[dE = - F \times dy = - ky \times dy \label{PEchange} \]
Ecuación de integración\ ref {PEChange} desde\(E = E\) y\(E = 0\)\(y = 0\) hacia\(y = y\)
\[\int_0^E dE = - k \int_0^y ydy \label{PEintegrals} \]
da la energía como
\[E = \frac{1}{2} k y^2 \label{PE} \]
La figura\(\PageIndex{4}b\) muestra la curva de energía potencial resultante, para la cual la energía potencial máxima es\(\frac{1}{2}kA^2\) cuando el peso está en su desplazamiento máximo. Tenga en cuenta que la curva de energía potencial es una parábola.
Frecuencia Vibracional
El oscilador armónico simple descrito anteriormente y mostrado en la Figura\(\PageIndex{4}\) vibra con una frecuencia\(\nu_0\), dada por la ecuación
\[\nu_0 = \frac{1}{2 \pi} \sqrt{\frac{k}{m}} \label{natfreq} \]
donde\(k\) es constante la fuerza del resorte y\(m\) es la masa del peso. Podemos extender esto a un resorte que conecta dos pesos entre sí sustituyendo la masa\(m\), la masa reducida del sistema,\(\mu\)
\[\mu = \frac{m_1 \times m_2}{m_1 + m_2} \label{redmass} \]
donde\(m_1\) y\(m_2\) son las masas de los dos pesos. Sustituyendo la ecuación\ ref {redmass} en la ecuación\ ref {natfreq} da
\[\nu_0 = \frac{1}{2 \pi} \sqrt{\frac{k}{\mu}} = \frac{1}{2 \pi} \sqrt{\frac{k(m_1 + m_2)}{m_1 \times m_2}} \label{natfreq2} \]
Si hacemos la suposición de que la Ecuación\ ref {natfreq2} se aplica a moléculas diatómicas simples, entonces podemos estimar la constante de fuerza del enlace\(k\),, midiendo su frecuencia vibracional.
Tratamiento Quantum de Vibraciones
Las ecuaciones\ ref {PE}\ y\ ref {natfreq2} se basan en un tratamiento mecánico clásico del oscilador armónico simple en el que cualquier desplazamiento, y, por lo tanto, cualquier energía es posible. Las vibraciones moleculares, sin embargo, se cuantifican; así
\[E = \left( v + \frac{1}{2} \right) \times h \times \frac{1}{2 \pi} \sqrt{\frac{k}{\mu}} = \left( v + \frac{1}{2} \right) h \nu_0 \label{quantizedE} \]
donde\(v\) está el número cuántico vibracional, que ha permitido valores de\(0, 1, 2, \dots\). La diferencia en energía,\(\Delta E\), entre dos niveles de energía vibratoria consecutivos cualesquiera es\(h \nu_0\). Como las transiciones permitidas en la mecánica cuántica se limitan a\(\Delta \nu = \pm 1\) y como la diferencia de energía se limita a\(\Delta E = h \nu_0\), cualquier modo particular de vibración debería dar lugar a un solo pico.
Comportamiento Anarmónico
El comportamiento ideal descrito en la última sección, en el que cada movimiento vibratorio que produce un cambio en el momento dipolar resulta en un solo pico, no se sostiene debido a una variedad de razones, incluyendo las interacciones culómbicas entre los átomos a medida que se acercan y alejan entre sí. Un resultado de estos comportamientos no ideales es que el valor\(\Delta E\) no permanece constante para todos los valores del número cuántico vibracional\(v\). Para valores mayores de\(v\), el valor de\(\Delta E\) se vuelve más pequeño y las transiciones donde\(\Delta v = \pm 2\) o\(\Delta v = \pm 3\) se hacen posibles dando lugar a lo que se llaman líneas armónicas a frecuencias que son\(2 \times\) o\(3 \times\) que para\(\nu_0\).
¿Por qué vemos más o menos picos vibratorios de lo esperado?
La figura\(\PageIndex{5}\) muestra el espectro IR para dióxido de carbono, CO 2, el cual consiste en tres cúmulos de picos ubicados aproximadamente a 670 cm —1, 2350 cm —1 y 3700 cm —1. Como el dióxido de carbono es una molécula lineal que consta de dos dobles enlaces carbono-oxígeno (O=C=O), tiene modos\(3 \times 3 - 5 = 9 - 5 = 4\) vibracionales. Entonces, ¿por qué vemos solo tres cúmulos de picos?
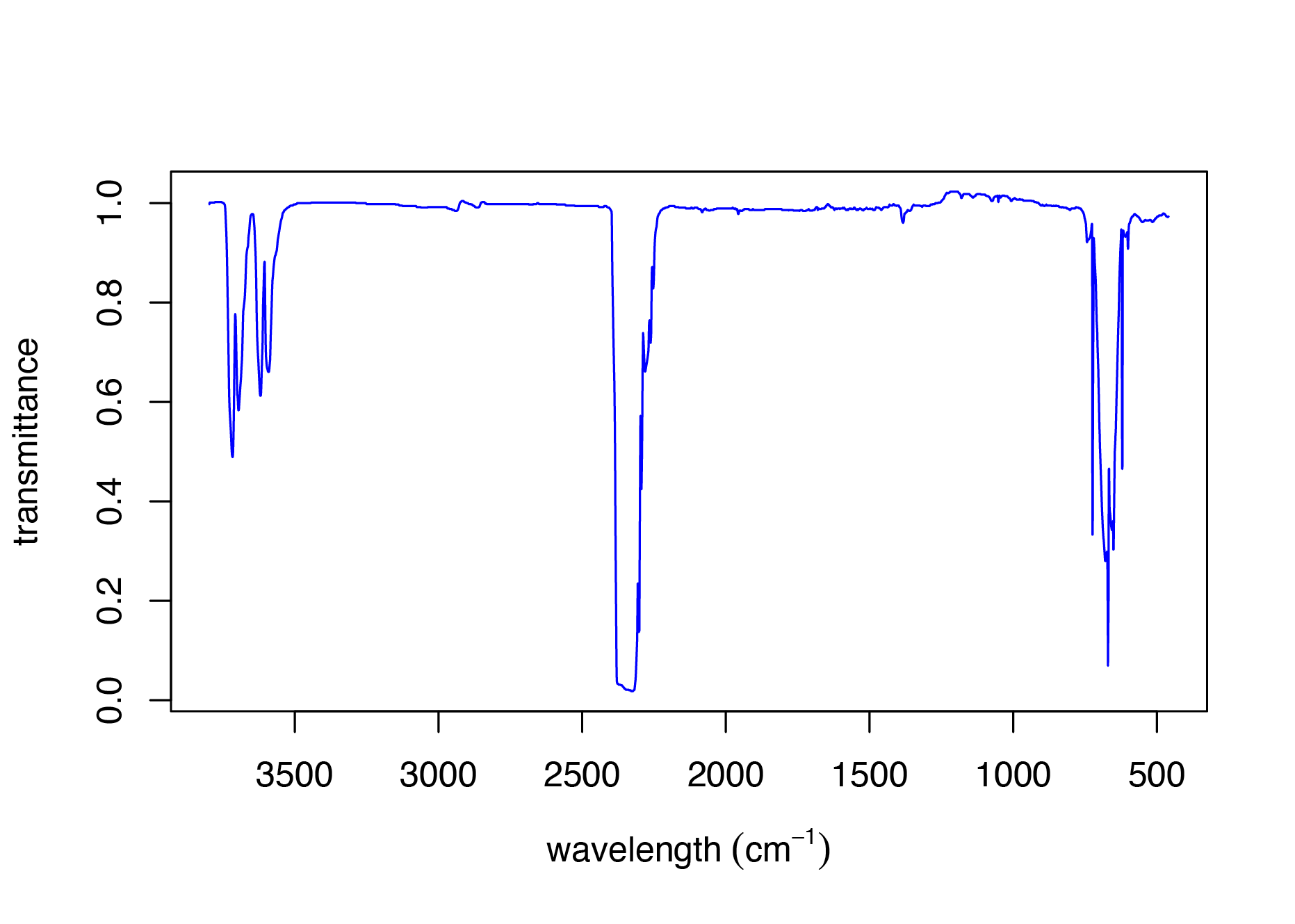
Uno de los requisitos para la absorción de la radiación infrarroja, es que el movimiento vibratorio debe resultar en un cambio en el momento dipolar. La figura\(\PageIndex{6}\) muestra los cuatro modos vibracionales para CO 2. De estos cuatro modos vibracionales, el estiramiento simétrico no da como resultado un cambio en el momento dipolar. Si bien esto parece explicar por qué vemos solo tres cúmulos de picos, un examen minucioso de los dos movimientos de flexión en Figura\(\PageIndex{6}\) debería convencerte de que son idénticos y, por lo tanto, aparecerán como un solo pico.
Entonces, ¿cuál es la fuente del cúmulo de picos alrededor de 3700 cm —1? En ocasiones la absorción de un solo fotón excita dos o más modos vibracionales. En este caso, el número de onda para esta banda de absorción es equivalente a la suma de los números de onda para el estiramiento asimétrico y los dos modos de flexión degenerados (2349 + 667 = 3016 cm —1, y 2349 + 667 + 667 = 3683 cm —1). A estas se les llama bandas de combinación.
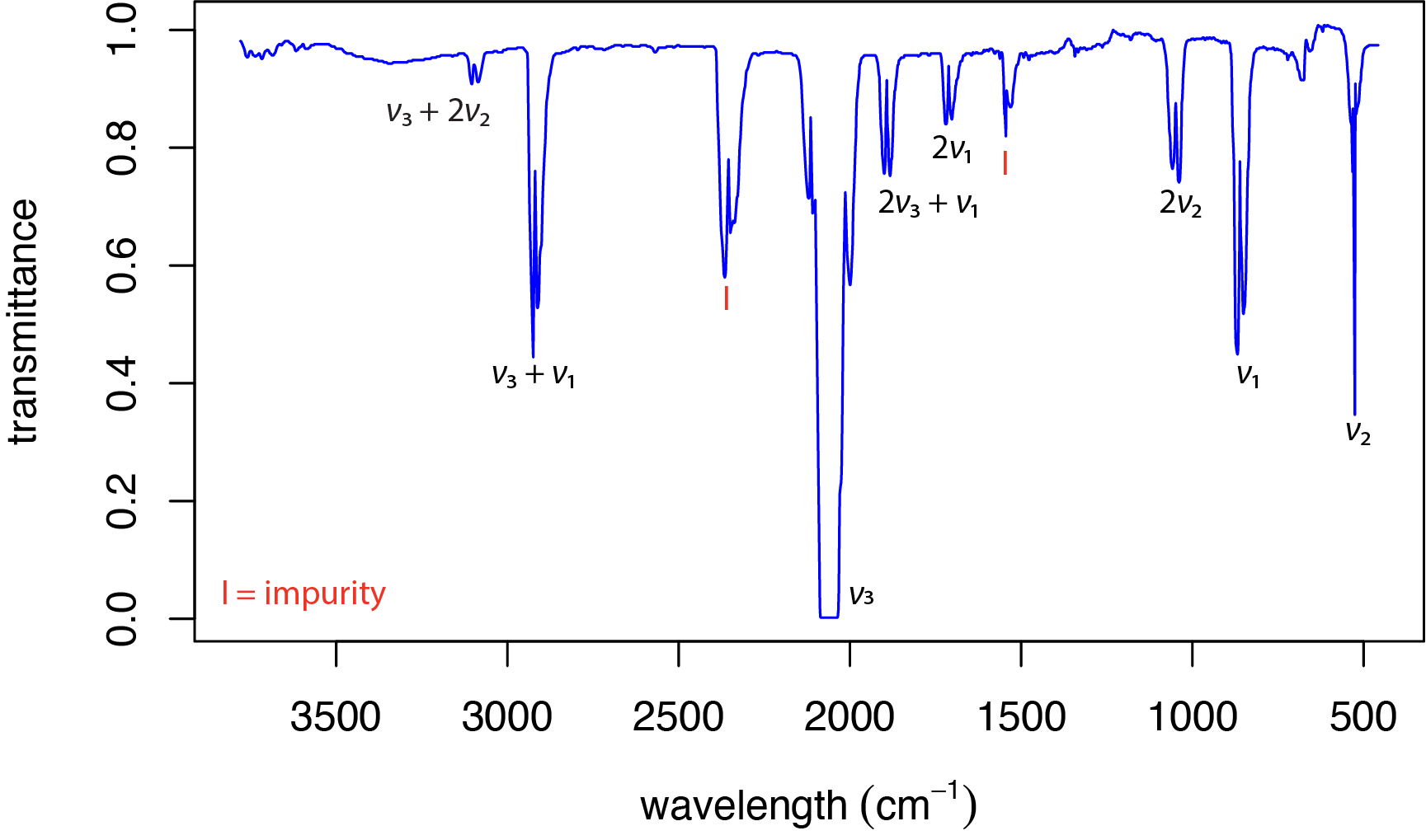
Otra fuente de picos adicionales son las bandas armónicas en las que\(\Delta v = \pm 2\) o\(\Delta v = \pm 3\). La figura\(\PageIndex{7}\) muestra el espectro IR para sulfuro de carbonilo, OCS, el cual es analago al CO 2 en el que uno de los oxígenos es reemplazado por azufre. El pico a 520 cm —1 es por sus dos movimientos de flexión degenerados y está etiquetado\(\nu_2\). El estiramiento asimétrico a 2062 cm —1\((\nu_3)\) y el tramo simétrico a 859 cm —1\((\nu_1)\) son las otras dos bandas de absorción fundamentales. Los picos restantes son armónicos, como el pico marcado\(2 \nu_2\) a 1040 cm —1, o bandas de combinación, como el pico marcado\(\nu_3 + \nu_1\) a 2921 cm —1. Muchos de los picos aparecen como dos picos; esto es el resultado de cambios en la energía rotacional también.