
1.7: Resolución estructural
- Page ID
- 77878

En el contexto de este capítulo, también se le invitará a visitar estas secciones...
- Métodos de cristalización para proteínas
- Simetría cristalina y simetría de difracción
- Condiciones para ausencias sistemáticas
- La función Patterson y el método Patterson
- La dispersión anómala
En capítulos anteriores, hemos visto cómo los rayos X interactúan con la materia estructurada periódicamente (cristales), y la pregunta implícita que hemos planteado de estos capítulos anteriores es:
¿Podemos “ver” la estructura interna de los cristales? , o en otras palabras,
¿Podemos “ver” los átomos y moléculas que construyen cristales?
¡La respuesta es definitivamente sí!
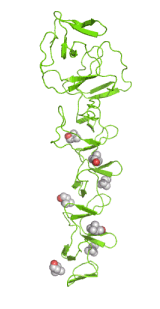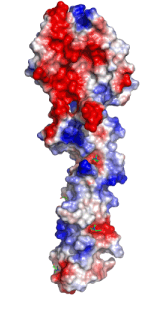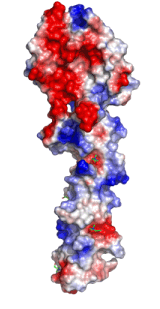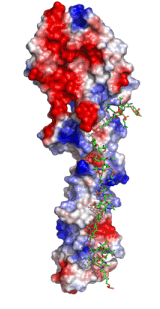

Izquierda: Estructura molecular de una enzima de superficie neumocócica
Centro: Empaque molecular en el cristal de un compuesto orgánico simple, mostrando su celda unitaria cristalográfica
Derecha: Detalles geométricos que muestran varias interacciones moleculares en un fragmento de la estructura molecular de una proteína
Como demuestran los ejemplos anteriores, la cristalografía puede mostrarnos las estructuras de estructuras moleculares muy grandes y complicadas (figura izquierda) y cómo las moléculas se agrupan en una estructura cristalina (figura central). También podemos ver cada detalle geométrico, así como los diferentes tipos de interacciones, entre moléculas o partes de ellas (figura derecha).
No obstante, para una mejor comprensión de los fundamentos en los que se basa esta respuesta, es necesario introducir algunos conceptos nuevos o refrescar algunos de los previamente vistos...
En capítulos anteriores hemos visto que los cristales representan la materia organizada y ordenada, consistente en asociaciones de átomos y/o moléculas, correspondientes a un estado natural de la misma con un mínimo de energía.
También sabemos que los cristales pueden describirse repitiendo unidades en las tres direcciones del espacio, y que a este espacio se le conoce como espacio directo o real. Estas unidades repetitivas se conocen como celdas unitarias (que también sirven como sistema de referencia para describir las posiciones atómicas). Este espacio directo o real, el mismo en el que vivimos, puede ser descrito por la densidad de electrones, ρ (xyz), una función definida en cada punto de la celda unitaria de coordenadas (xyz), donde además operan elementos de simetría que repiten átomos y moléculas dentro de la célula.

Celda unitaria (izquierda) cuyo apilamiento tridimensional construye un cristal (derecha)

Los motivos (átomos, iones o moléculas) se repiten por operadores de simetría dentro de la celda unitaria.
Las celdas unitarias se apilan en tres dimensiones, siguiendo las reglas de la celosía, construyendo el cristal.
También hemos aprendido que los rayos X interactúan con los electrones de los átomos en los cristales, dando como resultado un patrón de difracción, también conocido como espacio recíproco, con las propiedades de una red ( retícula recíproca) con una cierta simetría, y donde también podemos definir una celda repetitiva (celda recíproca). Los “puntos” de esta red recíproca contienen la información sobre la intensidad de difracción.


Izquierda: Interacción entre dos ondas dispersas por electrones. Las ondas resultantes muestran áreas de oscuridad (interferencia destructiva), dependiendo del ángulo considerado. Imagen tomada originalmente de physics-animations.com.
Derecha: Una de las cientos de imágenes de difracción de un cristal de proteína. Los puntos negros en la imagen son el resultado de la dispersión cooperativa (difracción) de los electrones de todos los átomos contenidos en el cristal.
A través de esta dispersión cooperativa (difracción), las ondas dispersas interactúan entre sí, produciendo un solo haz difractado en cada dirección del espacio, de manera que, dependiendo de las diferencias de fase (avance o retraso) entre las ondas dispersas individuales, suman o restar, como se muestra en las dos figuras siguientes:
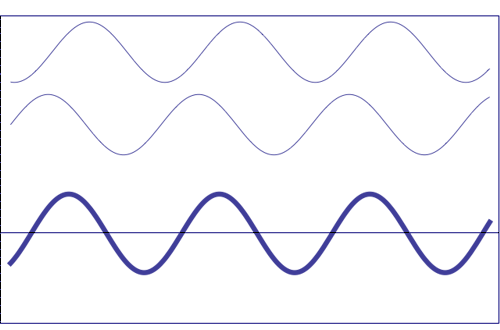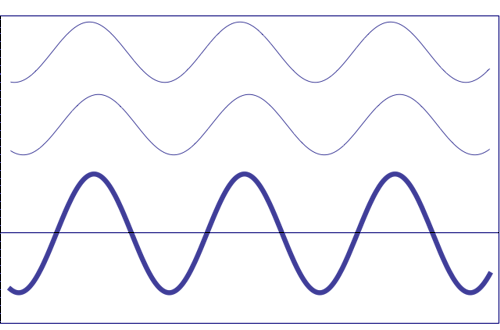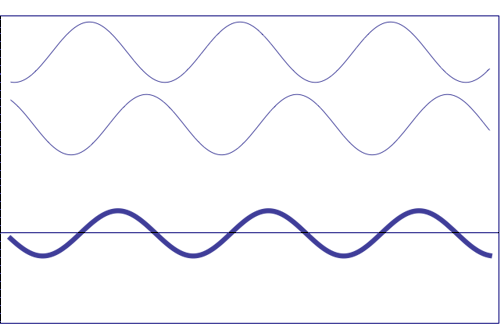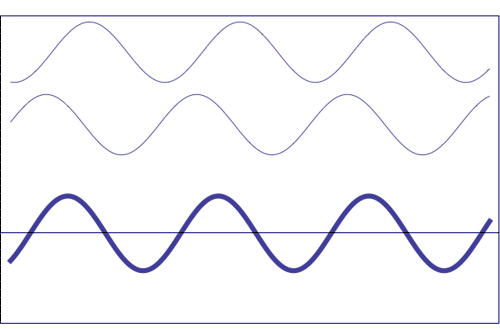
Interferencia de dos ondas con la misma amplitud y frecuencia (animación tomada de la Universidad Estatal de Pensilvania)

Composición de dos ondas dispersas. A = amplitud resultante; I = intensidad resultante (~ A 2)
(a) totalmente en fase (el efecto total es la suma de ambas ondas)
b) con cierta diferencia de fase (suman, pero no totalmente)
(c) fuera de fase (la amplitud resultante es cero)
Entre los dos espacios mencionados (directo y recíproco) existe una relación holística (cada detalle de uno de los espacios afecta al conjunto del otro, y el vicio versa). Matemáticamente hablando esta relación es una transformada de Fourier que no puede resolverse directamente, ya que el experimento de difracción no permite conocer una de las magnitudes fundamentales de la ecuación, las fases relativas (Φ) de los haces de difracción.


Izquierda: Relación holística entre el espacio directo (izquierda) y el espacio recíproco (derecha). Cada detalle del espacio directo (izquierda) depende de la información total contenida en el espacio recíproco (derecha), y viceversa... Cada detalle del espacio recíproco (derecha) depende de la información total contenida en el espacio directo (izquierda).
Derecha: Representación gráfica de la desfase entre dos ondas. Fase relativa entre olas
El siguiente diagrama, con la ayuda del siguiente párrafo, resume lo que implica la resolución de una estructura cristalina mediante difracción de rayos X...
Los átomos, iones y moléculas se empaquetan en unidades (células elementales) que se apilan en tres dimensiones para formar un cristal en el espacio que llamamos espacio directo o real. Los efectos de difracción del cristal se pueden representar como puntos de un espacio matemático reticular que llamamos la red recíproca. Las intensidades de difracción, es decir, el ennegrecimiento de estos puntos de la red recíproca, representan los módulos de algunas cantidades vectoriales fundamentales, a las que llamamos factores de estructura. Si llegamos a conocer no sólo los módulos de estos vectores (las intensidades), sino sus orientaciones relativas (es decir, sus fases relativas), podremos obtener el valor de la función de densidad electrónica en cada punto de la celda elemental, proporcionando así las posiciones de los átomos que componen el cristal.

Esquema sobre conceptos cristalográficos básicos: espacios directos y recíprocos. El problema es obtener información del lado izquierdo (espacio directo) del experimento de difracción (espacio recíproco).
DENSIDAD DE ELECTRONES
Para conocer (o ver) la estructura interna de un cristal tenemos que resolver una función matemática conocida como la “densidad electrónica”; una función que se define en cada punto de la celda unitaria (un concepto básico de la estructura cristalina introducido en otro capítulo).
La función de densidad electrónica, representada por la letra ρ, tiene que resolverse en cada punto dentro de la celda unitaria dada por las coordenadas (x, y, z), referidos a los ejes de las celdas unitarias. En aquellos puntos donde esta función toma valores máximos (estimados en términos de electrones por Angstrom cúbico) es donde se localizan los átomos. Eso quiere decir que si somos capaces de calcular esta función, “veremos” la estructura atómica del cristal.


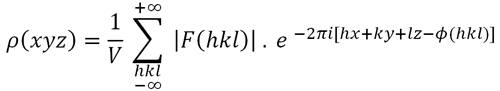
Fórmula 1. Función que define la densidad electrónica en un punto de la celda unitaria dada por las coordenadas (x, y, z)
- F (hkl) representa los haces difractados resultantes de todos los átomos contenidos en la celda unitaria en una dirección dada. Estas magnitudes (en realidad ondas), una por cada haz difractado, se conocen como factores de estructura. Sus módulos están directamente relacionados con las intensidades difractadas.
- h, k, l son los índices Miller de los haces difractados (los puntos recíprocos) y Φ (hkl) representan las fases de los factores de estructura. V representa el volumen de la celda unitaria. La función tiene limitaciones debido a la medida en que se observa el patrón de difracción. El número de factores estructurales observados es finito, por lo que la síntesis solo será aproximada y puede mostrar algunos efectos de truncamiento.

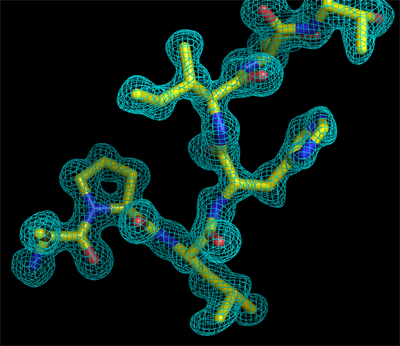
Izquierda: Apariencia de una zona del mapa de densidad electrónica de un cristal de proteína, antes de que se interprete.
Derecha: El mismo mapa de densidad electrónica después de su interpretación en términos de un fragmento peptídico.
La ecuación anterior (Fórmula 1) representa la transformada de Fourier entre el espacio real o directo (donde están los átomos, representada por la función ρ ) y el espacio recíproco (el patrón de rayos X) representado por las amplitudes de los factores de estructura y sus fases. La Fórmula 1 también muestra el carácter holístico de la difracción, ya que para calcular el valor de la densidad de electrones en un solo punto de coordenadas (xyz) es necesario utilizar el contribuciones de todos los factores de estructura producidos por la difracción cristalina.

Los factores de estructura F (hkl) son ondas y por lo tanto pueden representarse como vectores por sus amplitudes, [F (hkl)] y fases Φ (hkl) medido en un origen común de fases.
Cuando la celda unitaria es centrosimétrica, para cada átomo en las coordenadas (xyz) hay una idéntica ubicada en (-x, -y, -z). Esto implica que la ley de Friedel sostiene F (h, k, l) = F (-h, -k, -l) y la expresión de la densidad electrónica (Fórmula 1) se simplifica, convirtiéndose en la Fórmula 1.1. Y las fases de los factores de estructura también se simplifican, llegando a ser 0° o 180°...
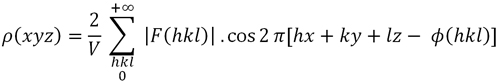
Fórmula 1.1. Función de densidad electrónica en un punto de coordenadas (x, y, z) en una celda unitaria centrosimétrica.
Es importante darse cuenta de que la cantidad y calidad de la información proporcionada por la función de densidad electrónica, ρ, es muy dependiente de la cantidad y calidad de los datos utilizados en la fórmula: los factores de estructura F ( hkl) (¡amplitudes y fases!). Veremos más adelante que las amplitudes de los factores estructurales se obtienen directamente del experimento de difracción.
Si tu navegador está habilitado para Java, como ejercicio práctico sobre las transformaciones de Fourier te recomendamos visitar los siguientes enlaces:
- o, mejor aún, el applet Java amablemente proporcionado por Nicholas Schöni y Gervais Chapui (École Polytechnique Fédérale de Lausanne, Suiza), que puedes descargar (libre de cualquier virus) desde el enlace que se muestra y ejecutar en tu propio computadora. Este applet calcula la transformada de Fourier de una función de densidad bidimensional ρ (x) produciendo la magnitud compleja G (S), el espacio recíproco. El applet también es capaz de calcular la transformada inversa de Fourier de G (S). La función de densidad puede ser periódica o no periódica. Numerosas herramientas, incluyendo herramientas de dibujo, se pueden aplicar para comprender el papel de las amplitudes y fases que son de particular importancia en los fenómenos de difracción. Como ilustración, se puede simular la función Patterson de una estructura periódica.
La expresión analítica de los factores estructurales, F (hkl), es simple e implica una nueva magnitud (¡j) llamada factor de dispersión atómica ( definido en un capítulo anterior) que toma en cuenta las diferentes potencias de dispersión con las que los electrones de los j átomos dispersan los rayos X:
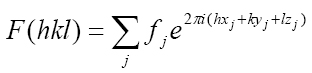
Fórmula 2. Factor de estructura para cada haz difractado. Esta ecuación es la transformada de Fourier de la densidad electrónica (Fórmula 1).
La expresión toma en cuenta los factores de dispersión ¡de todos los j átomos contenidos en la celda unitaria cristalina.
Desde el punto de vista experimental, es relativamente sencillo medir las amplitudes [F (hkl)] de todas las ondas difractadas producidas por un cristal. Solo necesitamos una fuente de rayos X, un solo cristal del material a estudiar y un detector apropiado. Con estas condiciones cumplidas podemos entonces medir las intensidades, I (hkl), de los haces difractados en términos de:
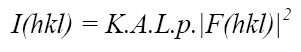
Fórmula 3. Relación entre la amplitud de los factores de estructura |F (hkl) | y sus intensidades I (hkl)
K es un factor que pone los factores de estructura experimental, (F rel), medidos en una escala relativa (que depende de la potencia de la fuente de rayos X, el tamaño del cristal, etc.) en una escala absoluta, que es digamos, la escala de los factores de estructura calculados (teóricos) (si pudiéramos conocerlos a partir de la estructura real, Fórmula 2 anterior). Como se desconoce la estructura en esta etapa, este factor puede evaluarse aproximadamente utilizando los datos experimentales mediante la denominada parcela Wilson.
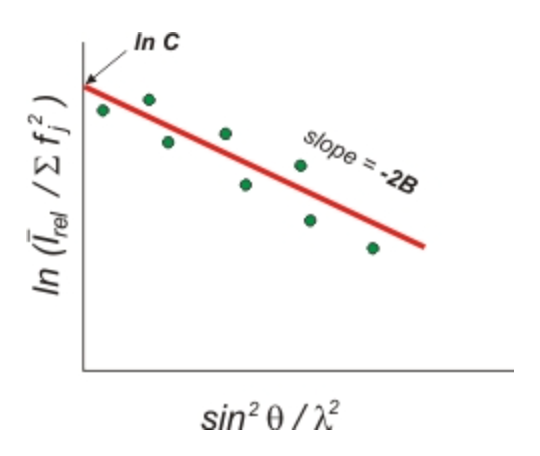
Parcela de Wilson
I rel representa la intensidad promedio (en una escala relativa) recogida en un intervalo dado de θ (el ángulo de Bragg); f j son los factores de dispersión atómica en ese rango angular, y λ es la longitud de onda de rayos X.
Al trazar las magnitudes mostradas en la figura izquierda (puntos verdes), se obtiene una línea recta de la que se puede derivar la siguiente información:
- El valor de la intersección del eje y es el logaritmo naperiano de C, una magnitud relacionada con el factor de escala K (= 1/√ C), descrito anteriormente.
- La pendiente es equivalente a -2B, donde B es el factor de vibración térmica atómica global isotrópica.
A es un factor de absorción, que puede estimarse a partir de las dimensiones y composición del cristal.
L es conocido como el factor Lorentz, responsable de corregir las diferentes velocidades angulares con las que los puntos recíprocos cruzan la superficie de la esfera de Ewald. Para los goniómetros de cuatro círculos este factor puede calcularse como 1/sin 2θ, donde θ es el ángulo de Bragg de las reflexiones.
p es el factor de polarización, que corrige el efecto de polarización del del haz incidente, y viene dado por la expresión (1+cos 2 2θ) /2, donde θ también representa el ángulo de Bragg del reflexiones (los puntos recíprocos).
EL PROBLEMA DE FASE
Sin embargo, para calcular la densidad de electrones (ρ (xyz) en la Fórmula 1, anterior), y por lo tanto conocer las posiciones atómicas dentro de la celda unitaria, también necesitan conocer las fases de los diferentes haces difractados (Φ (hkl) en la Fórmula 1 anterior). Pero, desafortunadamente, esta valiosa información se pierde durante el experimento de difracción (¡no hay técnica experimental disponible para medir las fases!) Así, debemos enfrentar el llamado problema de fase si queremos resolver la Fórmula 1.
El problema de fase se puede entender muy fácilmente si comparamos el experimento de difracción (como procedimiento para ver la estructura interna de los cristales) con un microscopio óptico convencional...

Ilustración sobre el problema de fase. Comparación entre un microscopio óptico y el microscopio de rayos X “imposible”. No hay lentes ópticas capaces de combinar rayos X difractados para producir una imagen zoom del contenido cristalino (átomos y moléculas).
En un microscopio óptico convencional la luz visible ilumina la muestra y los haces dispersos pueden recombinarse (con intensidad y fase) utilizando un sistema de lentes, dando lugar a una imagen ampliada de la muestra bajo observación.
En lo que podríamos llamar el microscopio de rayos X imposible (el proceso de visualización dentro de los cristales para localizar las posiciones atómicas), la luz visible es reemplazada por rayos X (con longitudes de onda cercanas a 1 Angstrom) y la muestra (el cristal) también dispersa esta “luz” (los rayos X). Sin embargo, no tenemos ningún sistema de lentes que puedan desempeñar el papel de las lentes ópticas, para recombinar las ondas difractadas proporcionándonos una “imagen” directa de la estructura interna del cristal. El experimento de difracción de rayos X solo nos da una imagen de la red recíproca del cristal en una placa fotográfica o detector. Lo único que podemos hacer en esta etapa es medir las posiciones e intensidades de las manchas recogidas en el detector. Estas intensidades son proporcionales a las amplitudes del factor de estructura, [F (hkl)].
Pero respecto a las fases, Φ (hkl), nada se puede concluir por el momento, impidiendo que obtengamos una solución directa de la función de densidad electrónica (Fórmula 1 anterior).
Por lo tanto, necesitamos algunas alternativas para recuperar los valores de fase, perdidos durante el experimento de difracción...
Resolución estructural
Una vez que se conoce y entiende el problema de fase, veamos ahora los pasos generales (ver el esquema a continuación) que debe enfrentar un cristalógrafo para resolver la estructura de un cristal y por lo tanto localizar las posiciones de los átomos, iones o moléculas contenidas en la celda unitaria...

Diagrama general que ilustra el proceso de resolución de estructuras moleculares y cristalinas por difracción de rayos X El proceso consiste en diferentes etapas que han sido tratadas previamente o que se describen a continuación:
- Conseguir un cristal adecuado para el experimento, con la calidad y tamaño adecuados. Algo relacionado se verá en otra sección.
- Obtención del patrón de difracción con la longitud de onda apropiada. Esto ha sido descrito en otro capítulo.
- Evaluar el patrón de difracción para obtener los parámetros de celosía (celda unitaria), simetría (grupo espacial) e intensidades de difracción.
- Resolviendo la función de densidad electrónica, obteniendo cualquier información sobre las fases de los haces difractados. Este es un punto clave para la resolución estructural que se discutirá a continuación.
- Construir un modelo estructural inicial para explicar los valores de la función de densidad electrónica y completar el modelo localizando las posiciones atómicas restantes. Esto se verá a continuación.
- Refinando el modelo, ajustando todas las posiciones atómicas para obtener el patrón de difracción calculado lo más similar posible al patrón de difracción experimental, y finalmente validar y mostrar el modelo estructural total obtenido. Esto se verá en otro capítulo.
Para que el estudio sea exitoso, se deben tomar en cuenta algunos aspectos importantes, tales como:
- El compuesto en estudio debe ser puro para ser cristalizado (si no ya, como en el caso de los minerales naturales).
- Los cristales se pueden obtener usando diferentes técnicas, desde el método más simple de evaporación o enfriamiento lento hasta el más complejo: difusión de vapor (o solvente), sublimación, convección, etc. Hay suficiente literatura disponible.. Consulte, por ejemplo, las páginas del LEC, Laboratorio de Estudios Cristalográficos, para obtener información adicional sobre técnicas de cristalización específicas. Para las proteínas, el procedimiento más utilizado se basa en experimentos de difusión de vapor, generalmente con la técnica de “gota colgante”, descrita en otra parte de estas páginas. En este sentido es muy relevante destacar los recientes avances introducidos en el campo de la nanocristalografía de proteínas de rayos X de femtosegundos, lo que significará un paso gigante para eliminar prácticamente la mayoría de las dificultades en el proceso de cristalización, y en particular para las proteínas (ver el pequeño párrafo dedicado al láser de electrones libres de rayos X).
- Si se obtienen cristales apropiados, se exponen a rayos X y se miden sus intensidades de difracción utilizando los métodos y equipos descritos en un capítulo anterior. Una cuidadosa evaluación de los datos nos proporcionará las dimensiones de la celda unitaria, la simetría y, directamente a partir de las intensidades, las amplitudes de los factores de estructura [F (hkl)]. De todos estos temas en esta etapa, el más difícil se refiere a la determinación de la simetría cristalina, cuestión clave para la resolución exitosa de la estructura. Para obtener simetría cristalina, un estudio visual del cristal no tendría sentido y por lo tanto debe deducirse de la simetría del patrón de difracción, como se indica en una sección específica de estas páginas.
- En esta etapa surge la pregunta sobre las fases desconocidas, Φ (hkl), por lo que de alguna manera deben ser evaluadas, como veremos a continuación...
- Si las fases evaluadas son correctas, la función de densidad electrónica ρ (xyz) mostrará una distribución de máximos (posiciones atómicas) consistente y significativa desde el punto de vista estereoquímico. Una vez que se conoce una estructura inicial, se deben llevar a cabo algunos pasos adicionales (construcción del modelo detallado, refinamiento matemático y validación). Esto nos llevará al llamado modelo final de la estructura.
Pero volvamos al tema más importante: ¿cómo resolvemos el problema de fase?
LA FUNCIÓN PATTERSON

La primera solución al problema de fase la introdujo Arthur Lindo Patterson (1902-1966).
Basando su trabajo en la incapacidad de resolver directamente la función de densidad electrónica (Fórmula 1 arriba o abajo), y después de su formación (bajo el matemático estadounidense Norbert Wiener) sobre la convolución de las transformaciones de Fourier, Patterson introdujo una nueva función P ( uvw) (Fórmula 4, abajo) en 1934. Esta fórmula, que define un nuevo espacio (el espacio Patterson), puede considerarse como el desarrollo único más importante en el análisis de la estructura cristalina desde el descubrimiento de rayos X por Röntgen en 1895 o rayos X difracción por Laue en 1914.
Su elegante fórmula, conocida como la función Patterson (Fórmula 4, a continuación), introduce una simplificación de la información contenida en la función de densidad electrónica. La función Patterson elimina el término que contiene las fases, y las amplitudes de los factores de estructura son reemplazadas por sus cuadrados. Es así una función que puede calcularse inmediatamente a partir de los datos experimentales disponibles (intensidades, que están relacionadas con las amplitudes de los factores de estructura). Formalmente, desde el punto de vista matemático, la función Patterson es equivalente a la convolución de la densidad electrónica (Fórmula 1, a continuación) con su inversa: ρ (x, y, z) * ρ (-x, -y, -z).
Fórmula 1. La función de densidad electrónica calculada en el punto de coordenadas (x, y, z).
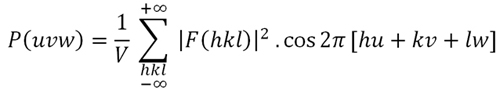
Fórmula 4. La función Patterson calculada en el punto (u, v, w) . Esto es una simplificación de la Fórmula 1 , ya que la suma se realiza en F 2 (hkl) y se supone que todas las fases son cero.
Parece obvio que después de omitir la información crucial contenida en las fases [Φ (hkl) en la Fórmula 1], la función Patterson ya no mostrará las posiciones directas de los átomos en la celda unitaria, como la función de densidad electrónica haría.De hecho, la función Patterson solo proporciona un mapa de vectores interatómicos (posiciones atómicas relativas), siendo la altura de sus máximos proporcional al número de electrones de los átomos implícitos. Veremos que esta característica significa una ventaja en la detección de las posiciones de los átomos “pesados” (con muchos electrones) en estructuras donde los átomos restantes tienen números atómicos más bajos. Una vez calculado el mapa de Patterson, tiene que ser interpretado correctamente (al menos parcialmente) para obtener las posiciones absolutas (x, y, z) de los átomos pesados dentro de la celda unitaria. Estas posiciones atómicas ahora se pueden utilizar para obtener las fases Φ (hkl) de los haces difractados invirtiendo la Fórmula 1 y, por lo tanto, esto permitirá el cálculo de la función de densidad electrónica ρ (xyz), pero éste será objeto de otra sección de estas páginas.
LOS MÉTODOS DIRECTOS
El problema de fase de los cristales formados por moléculas pequeñas y medianas fue resuelto satisfactoriamente por varios autores a lo largo del siglo XX con especial mención a Jerome Karle (1918-2013) y Herbert A. Hauptmann (1917-2011), quien compartió el Premio Nobel de Química en 1985 (sin olvidar el papel de Isabella Karle, 1921-2017). La metodología introducida por estos autores, conocida como los métodos directos, generalmente explota restricciones o correlaciones estadísticas entre las fases de diferentes componentes de Fourier.



Izquierda: Herbert A. Hauptman (1917-2011)
Centro: Jerome Karle (1918-2013)
Derecha: Isabella Karle (1921-2017)
La atomicidad de las moléculas, y el hecho de que la función de densidad electrónica debe ser cero o positiva en cualquier punto de la celda unitaria, crea ciertas limitaciones en la distribución de fases asociadas a los factores estructurales. En este contexto, los métodos directos establecen sistemas de ecuaciones que utilizan las intensidades de los haces difractados para describir estas limitaciones. La resolución de estos sistemas de ecuaciones proporciona información directa sobre la distribución de fases. Sin embargo, dado que la validez de cada una de estas ecuaciones se establece en términos de probabilidad, es necesario contar con un gran número de ecuaciones para sobredeterminar los valores de fase de las incógnitas (fases Φ (hkl)).

Los métodos directos utilizan ecuaciones que relacionan la fase de una reflexión (hkl) con las fases de otras reflexiones vecinas (h', k', l' y h-h', k-k', l-l'), asumiendo que estas relaciones son "probablemente verdaderas" (P )...
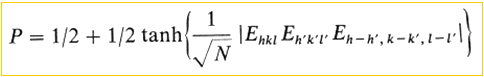
donde E hkl, E h'k'l' y E h-h', k-k', l-l' son los llamados “factores de estructura normalizados”, es decir, factores de estructura corregidos para el movimiento térmico, llevados a un absoluto escala y asumiendo que las estructuras están hechas de átomos puntuales. En otras palabras, la normalización del factor de estructura convierte los valores medidos de |F| en coeficientes de “átomos puntuales en reposo” conocidos como valores |E|.
En la actualidad, los métodos directos son los preferidos para los factores de estructura de fase producidos por moléculas pequeñas o medianas que tienen hasta 100 átomos en la unidad asimétrica. Sin embargo, generalmente no son factibles por sí mismos para moléculas más grandes como las proteínas. El lector interesado debe buscar una excelente introducción a los métodos directos a través de este enlace ofrecido por la Unión Internacional de Cristalografía.
Métodos de resolución estructural para macromoléculas
Para cristales compuestos por moléculas grandes, como proteínas y enzimas, el problema de fase puede resolverse exitosamente con tres métodos principales, dependiendo del caso:
(i) introducir átomos en la estructura con alto poder de dispersión. Esta metodología, conocida como MIR (Reemplazo Isomorfo Múltiple), se basa en el método de Patterson.
(ii) introducir átomos que dispersan los rayos X de manera anómala, también conocido como MAD (Difracción anómala de longitud de onda múltiple), y
(iii) mediante el método conocido como MR (Reemplazo Molecular), el cual utiliza la estructura previamente conocida de una proteína similar.
MIR (M ultiple I somorfa R eplacement)
Esta técnica, basada en el método Patterson, fue introducida por David Harker, pero fue aplicada con éxito por primera vez por Max F. Perutz y John C. Kendrew quienes recibieron el Premio Nobel en Química en 1962, por resolver la primera estructura de una proteína, la hemoglobina.



Izquierda: David Harker (1906-1991)
Centro: Max Ferdinand Perutz (1914-2002)
Derecha: John Cowdery Kendrew (1917-1997)
El método MIR se aplica después de introducir átomos “pesados” (dispersores grandes) en la estructura cristalina. Sin embargo, la dificultad de esta metodología radica en el hecho de que los átomos pesados no deben afectar la formación cristalina o las dimensiones de las células unitarias en comparación con su forma nativa, por lo tanto, deben ser isomórficos
Este método se lleva a cabo remojando el cristal de la muestra a analizar con una solución de átomos pesados o por co-cristalización con el átomo pesado, con la esperanza de que los átomos pesados pasen por los canales de la estructura cristalina y permanezcan unidos a cadenas laterales de aminoácidos con la capacidad de coordinarse átomos metálicos (por ejemplo, grupos SH de cisteína). En el caso de las metaloproteínas, se pueden reemplazar sus metales endógenos por otros más pesados (por ejemplo Zn por Hg, Ca por Sm, etc.).
Los átomos pesados (con un gran número de electrones) muestran una mayor potencia de dispersión que los átomos normales de una proteína (C, H, N, O y S), y por lo tanto cambian apreciablemente las intensidades del patrón de difracción cuando se comparan con la proteína nativa. Estas diferencias de intensidad entre los dos espectros (estructuras pesadas y nativas) se utilizan para calcular un mapa de vectores interatómicos entre las posiciones de los átomos pesados (mapa de Patterson), a partir del cual es relativamente fácil determinar su coordenadas dentro de la celda unitaria.

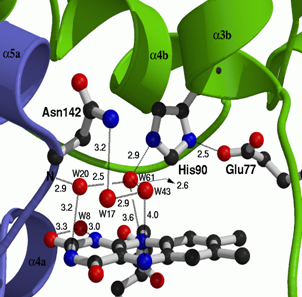
Esquema de una función Patterson derivada de un cristal que contiene tres átomos en la celda unitaria. Para obtener esta función gráficamente a partir de una estructura cristalina conocida (figura izquierda) se trazan todos los vectores interatómicos posibles (figura central). Estos vectores se mueven entonces paralelos a sí mismos al origen de la celda unitaria Patterson (figura derecha). La función calculada mostrará valores máximos al final de estos vectores, cuyas alturas son proporcionales al producto de los números atómicos de los átomos involucrados. Las posiciones en estos máximos (con coordenadas u, v, w) representan las diferencias entre las coordenadas de cada par de átomos en el cristal, es decir, u=x 1 -x 2, v=y 1 -y 2, w=z 1 -z 2.
Con las posiciones conocidas de los átomos pesados, los factores de estructura ahora se calculan usando la Fórmula 2 (ver también el diagrama a continuación), es decir sus amplitudes |F c (hkl) | y fases Φ c (hkl), donde el subíndice c significa “calculado”. Al usar la Fórmula 1, se calcula ahora un mapa de densidad electrónica, ρ (xyz), utilizando las amplitudes de los factores de estructura observados en el experimento, |F o (hkl) | (que contiene la contribución de toda la estructura) combinado con las fases calculadas Φ c (hkl). Si estas fases son lo suficientemente buenas, el mapa de densidad electrónica calculado mostrará no solo los átomos pesados conocidos, sino que también dará información adicional sobre otras posiciones atómicas (ver diagrama a continuación).
En resumen, los pasos de la metodología MIR son:
- Preparar uno o varios derivados de átomos pesados que deben ser isomórficos con la proteína nativa. Se realiza una primera prueba de isomorfismo en términos de los parámetros de la celda unitaria.
- Recopilar datos de difracción de derivados de átomos nativos y pesados.
- Aplica el método Patterson para obtener las posiciones de los átomos pesados.
- Refine estas posiciones atómicas y calcule las fases para todos los haces difractados.
- Obtener un mapa de densidad electrónica con esas fases calculadas.
MAD (M ulti-longitud de onda A nomalous D iffraction)
Los cambios en la intensidad de los datos de difracción producidos por la introducción de átomos pesados en los cristales de proteína pueden considerarse como una modificación química del experimento de difracción. De igual manera, podemos provocar cambios en la intensidad de difracción modificando las propiedades físicas de los átomos. Así, si la radiación de rayos X incidente tiene una frecuencia cercana a la frecuencia de vibración natural de los electrones en un átomo dado, el átomo se comporta como un “dispersor anómalo”. Esto produce algunos cambios en el factor de dispersión atómica, ¡j (ver Fórmula 2), de manera que su expresión es modificada por dos términos, ƒ' y ƒ” que dan cuenta de sus componentes reales e imaginarios, respectivamente. Para los átomos que se comportan de manera anómala, su factor de dispersión viene dado por la expresión que se muestra a continuación (Fórmula 5).
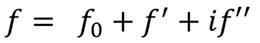
Fórmula 5. Ante la presencia de dispersión anómala, el factor de dispersión atómica, ¡0 , tiene que ser modificado agregando dos nuevos términos, una parte real y otra imaginaria.
El lector avanzado también debe leer la sección sobre el fenómeno de dispersión anómala.
Las correcciones ¡'y ¡¡” vs. energía de rayos X (ver a continuación para el caso de Cu Kα) pueden calcularse tomando en cuenta algunas consideraciones teóricas...

Componentes reales e imaginarios del factor de dispersión de Selenio vs. la energía de los rayos X incidentes. La línea vertical indica la longitud de onda para CUKα.
Para los valores de energía de rayos X donde existe resonancia, ƒ' aumenta dramáticamente, mientras que el valor de ¡” disminuye. Esto tiene importancia práctica considerando que muchos átomos pesados utilizados en cristalografía muestran picos de absorción a energías (longitudes de onda) que se pueden obtener fácilmente con radiación de sincrotrón. Los datos de difracción recogidos en estas condiciones mostrarán un componente normal, principalmente debido a los átomos de luz (nitrógeno, carbono e hidrógeno), y una parte anómala producida por los átomos pesados, lo que producirá un cambio global en la fase de cada reflexión. Todo esto lleva a un cambio de intensidad entre aquellas reflexiones conocidas como pares de Friedel (pares de reflexiones que en condiciones normales deberían tener las mismas amplitudes y fases idénticas, pero con signos opuestos). El cambio detectable de intensidad entre estos pares de reflexión (pares de Friedel) es lo que llamamos difracción anómala.
El método MAD, desarrollado por Hendrickson y Kahn, implica la medición de datos de difracción del cristal proteico (que contiene un dispersor anómalo fuerte) usando radiaciones de rayos X con diferentes energías (longitudes de onda): una que maximiza ƒ' ', otra que minimiza ƒ' y una tercera medición a un valor energético distinto de estos dos. Combinando estos conjuntos de datos de difracciones, y analizando específicamente las diferencias entre ellos, es posible calcular la distribución de amplitudes y fases generadas por los dispersores anómalos. El uso posterior de las fases generadas por estos dispersores anómalos, como primera aproximación, puede emplearse para calcular un mapa de densidad electrónica para toda la proteína.
En general, actualmente no hay necesidad de introducir átomos individuales como dispersores anómalos en los cristales proteicos. Es relativamente fácil obtener proteínas recombinantes en las que los residuos de metionina son reemplazados por selenio-metionina. Los átomos de selenio (e incluso azufre) de la metionina (o cisteína), se comportan como dispersores anómalos adecuados para llevar a cabo un experimento de MAD.
El método MAD presenta algunas ventajas frente a la técnica MIR:
- Como la técnica MAD utiliza datos recolectados de un solo cristal, no se aplican los problemas derivados de la falta de isomorfismo, común en el método MIR.
- Mientras que en ausencia de dispersión anómala, el factor de dispersión atómica (¡0) disminuye drásticamente con el ángulo de dispersión, su componente anómalo (ƒ' + i ¡"”) es independiente de ese ángulo, de manera que esta señal relativa aumenta a una mayor resolución del espectro, es decir, en ángulos altos de Bragg. Por lo tanto, las estimaciones de fases por MAD son generalmente mejores a alta resolución. Por el contrario, con el método MIR, la falta de isomorfismo es mayor en ángulos de alta resolución y por lo tanto las intensidades de alta resolución (> 3.5 Angstrom) no son adecuadas para la fase.
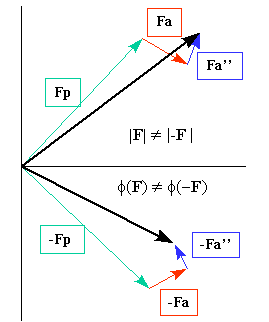
Diagrama de Argand que muestra la contribución de dispersión de un dispersor anómalo en una matriz de dispersores normales. Este efecto implica que la ley de Friedel falla. Imagen tomada de "Cristalografía 101”.
- Fp representa la contribución de los dispersores normales al factor de estructura (de índices hkl).
- Fa y Fa” representan las partes real (¡0 + ƒ') e imaginaria (¡"”), respectivamente, del factor de dispersión de los dispersores anómalos.
- -Fp, -Fa y -Fa” representan lo mismo que Fp, Fa y Fa”, pero para la reflexión con índices -h, -k, -l.
El comportamiento anómalo del factor de dispersión atómica solo produce pequeñas diferencias entre las intensidades (y por tanto entre las amplitudes de los factores de estructura) de las reflexiones que están relacionadas por un centro de simetría o un plano espejo (como por ejemplo, I (h, k, l) vs. I (-h, -k, -l), o I (h, k, l) vs. I (h, -k, l). Por lo tanto, para estimar estas pequeñas diferencias entre las intensidades experimentales, se deben tomar en cuenta precauciones adicionales. Por lo tanto, se recomienda que las reflexiones que se espera que muestren estas diferencias se recojan en la misma imagen de difracción, o alternativamente, después de cada imagen recolectada, rote el cristal 180 grados y recoja una nueva imagen. Además, dado que los cambios en ƒ' y ƒ” ocurren por variaciones mínimas de energía de rayos X, es necesario tener un buen control de los valores de energía (longitudes de onda). Por lo tanto, es esencial utilizar una instalación de radiación sincrotrón, donde las longitudes de onda se puedan afinar fácilmente.
El lector avanzado también debe echar un vistazo a las páginas web sobre dispersión anómala, preparadas por Bernhard Rupp, así como el resumen práctico elaborado por Georg M. Sheldrick.
MR (colocación R olecular M)
Si conocemos el modelo estructural de una proteína con una secuencia de aminoácidos homóloga, el problema de fase puede resolverse utilizando la metodología conocida como reemplazo molecular (MR). La estructura conocida de la proteína homóloga es considerada como la proteína a determinar y sirve como primer modelo para ser posteriormente refinada. Este procedimiento se basa obviamente en la observación de que las proteínas con secuencias peptídicas similares muestran un plegamiento muy similar. El problema en este caso es transferir la estructura molecular de la proteína conocida de su propia estructura cristalina a un nuevo empaquetamiento cristalino de la proteína con una estructura desconocida. El posicionamiento de la molécula conocida en la célula unitaria de la proteína desconocida requiere determinar su correcta orientación y posición dentro de la célula unitaria. Ambas operaciones, rotación y traslación, se calculan utilizando las llamadas funciones de rotación y traslación (ver abajo).

Esquema del método de reemplazo molecular (RM).
La molécula con estructura conocida (A ) se gira a través de la operación [R] y se desplaza a través de T para llevarla sobre la posición de lo desconocido molécula (A').
La función de rotación. Si consideramos el caso de dos moléculas idénticas, orientadas de manera diferente, entonces la función Patterson contendrá tres conjuntos de vectores. El primero contendrá los vectores Patterson de una de las moléculas, es decir, todos los vectores interatómicos dentro de la molécula uno (también llamados vectores propios). El segundo conjunto contendrá los mismos vectores pero para la segunda molécula, idénticos a la primera, pero rotados debido a su diferente orientación. El tercer conjunto de vectores serán los vectores cruzados interatómicos entre las dos moléculas. Mientras que los vectores propios están confinados al volumen ocupado por la molécula, los vectores cruzados se extenderán más allá de este límite. Si ambas moléculas (conocidas y desconocidas) son muy similares en estructura, la función de rotación R (α, β, γ) intentaría que los vectores Patterson de una de las moléculas coincidan con los de la otra, hasta que estén de acuerdo. Esta metodología fue descrita por primera vez por Rossman y Blow.
R (α, β, γ) = ∫ u P 1 (u) x P 2 (u r) du
Fórmula 6. Función de rotación
P 1 es la función Patterson y P 2 es la función Patterson girada, donde u es el volumen de el mapa de Patterson, donde se calculan los vectores interatómicos.
La calidad de las soluciones de estas funciones se expresa por el coeficiente de correlación entre ambas funciones de Patterson: la experimental y la calculada (con la proteína conocida). Un alto coeficiente de correlación entre estas funciones equivale a una buena concordancia entre el patrón de difracción experimental y el patrón de difracción calculado con la estructura proteica conocida. Una vez que la estructura proteica conocida está correctamente orientada y traducida (dentro de la célula unitaria de la proteína desconocida), se calcula un mapa de densidad electrónica utilizando estas posiciones atómicas y los factores de estructura experimental. Vale la pena consultar el artículo publicado sobre esta metodología por Eleanor Dodson.
Probablemente sea valioso para el lector avanzado consultar un bonito artículo que, a pesar de haber sido publicado en 2010, no ha perdido su validez en relación con la descripción de las diferentes metodologías para la determinación de las fases relativas de los haces de difracción.
COMPLETAR LA ESTRUCTURA
Todos estos métodos (Patterson, métodos directos, MIR, MAD, MR) proporcionan (directa o indirectamente) conocimiento sobre las fases aproximadas que deben actualizarse. Como se indicó anteriormente, las fases iniciales calculadas, Φ c (hkl), junto con las amplitudes experimentales observadas, |F o (hkl) |, nos permiten calcular un mapa de densidad electrónica, también aproximado, sobre el cual podemos construir el modelo estructural. El proceso general se resume en el diagrama cíclico que se muestra a continuación.
Las fases iniciales, Φ c (hkl), se combinan con las amplitudes de los factores estructurales experimentales (observados), |F o (hkl) |, y un electrón se calcula el mapa de densidad (se muestra en la parte inferior del esquema). Alternativamente, si los datos iniciales conocidos son las coordenadas (xyz) de algunos átomos, proporcionarán las fases iniciales (mostradas en la parte superior del esquema), y así sucesivamente de manera cíclica hasta que el proceso no produzca ninguna nueva información.

Esquema que muestra un proceso cíclico para calcular mapas de densidad electrónica ρ (xyz) que producen más información estructural.
A partir de varias posiciones atómicas conocidas siempre podemos calcular los factores de estructura: sus amplitudes, |Fc (hkl) |, y sus fases, Φc (hkl), como se muestra en la parte superior del esquema. Obviamente, las amplitudes calculadas pueden ser rechazadas, porque se calculan a partir de una estructura parcial y las experimentales representan la estructura total y real. Por lo tanto, el mapa de densidad electrónica (mostrado en la parte inferior del esquema) se calcula con las amplitudes experimentales (u observadas), |Fo (hkl) |, y las fases calculadas, Φc (hkl). Esta función se evalúa ahora en términos de posibles nuevas posiciones atómicas que se suman a las previamente conocidas, y el ciclo repetido. Históricamente este proceso se conocía como “síntesis sucesiva de Fourier”, debido a que la densidad electrónica se calcula en términos de una suma de Fourier.
En cualquier caso, a partir de posiciones atómicas o directamente de fases, si la información es correcta, la función de densidad de electrones será interpretable y contendrá información adicional (nuevas coordenadas atómicas) que pueden ser inyectadas en el procedimiento cíclico mostrado anteriormente hasta la finalización de la estructura, que es decir hasta que la función calculada ρ (xyz) no muestre cambios desde el último cálculo.
Los átomos más ligeros de la estructura (aquellos con menor número atómico, es decir, generalmente átomos de hidrógeno) son los más difíciles de encontrar en un mapa de densidad electrónica. Su poder de dispersión está casi oscurecido por la dispersión de los átomos restantes. Por esta razón, la ubicación de los átomos de H se realiza normalmente a través de una función de densidad electrónica algo modificada (la densidad electrónica de diferencia), cuyos coeficientes son las diferencias entre los factores de estructura observados y calculados del modelo conocido hasta ahora:
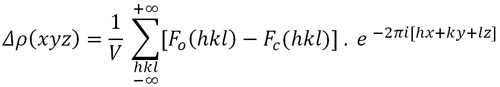
Fórmula 7. Función de densidad electrónica “diferencia”
En la práctica, si el modelo estructural obtenido es lo suficientemente bueno, si el experimento proporcionó factores de estructura precisos, y no hay errores específicos como la absorción de rayos X, el mapa de diferencias Δρ contendrá suficiente señal (máximos) donde se puedan ubicar los átomos de H. Adicionalmente, para obtener una señal mejorada a partir de la dispersión de los átomos de luz, esta función generalmente se calcula con los factores de estructura que aparecen solo en ángulos de difracción más bajos, generalmente con los que aparecen en sen θ/λ < 0.4, es decir, usando la región en la que los factores de dispersión de los hidrógenos siguen siendo “visibles”.