
3.1: Principios de Cromatografía de Gases
- Page ID
- 71191

Archer J.P. Martin (Figura\(\PageIndex{1}\)) y Anthony T. James (Figura\(\PageIndex{2}\)) introdujeron la cromatografía de reparto líquido-gas en 1950 en la reunión de la Biochemical Society celebrada en Londres, unos meses antes de presentar tres artículos fundamentales al Biochemical Journal. Fue este trabajo el que proporcionó las bases para el desarrollo de la cromatografía de gases. De hecho, Martin imaginó la cromatografía de gases casi diez años antes, mientras trabajaba con R. L. M. Synge (Figura\(\PageIndex{3}\)) en cromatografía de partición. Martin y Synge, quienes fueron galardonados con el premio Nobel de Química en 1941, sugirieron que la separación de compuestos volátiles podría lograrse usando un vapor como fase móvil en lugar de un líquido.



La cromatografía de gases rápidamente ganó aceptación general porque se introdujo en el momento en que se requerían mejores controles analíticos en las industrias petroquímicas, y se necesitaban nuevas técnicas para superar las limitaciones de los métodos de laboratorio antiguos. Hoy en día, la cromatografía de gases es una técnica madura, ampliamente utilizada a nivel mundial para el análisis de casi todos los tipos de compuestos orgánicos, incluso aquellos que no son volátiles en su estado original pero que pueden convertirse en derivados volátiles.
El Proceso Cromatográfico
La cromatografía de gases es una técnica de separación en la que los componentes de una muestra se dividen entre dos fases:
- La fase estacionaria.
- La fase gaseosa móvil.
Según el estado de la fase estacionaria, la cromatografía de gases puede clasificarse en cromatografía gas-sólido (GSC), donde la fase estacionaria es sólida, y cromatografía gas-líquido (GLC) que utiliza un líquido como fase estacionaria. GLC es en gran medida más ampliamente utilizado que GSC.
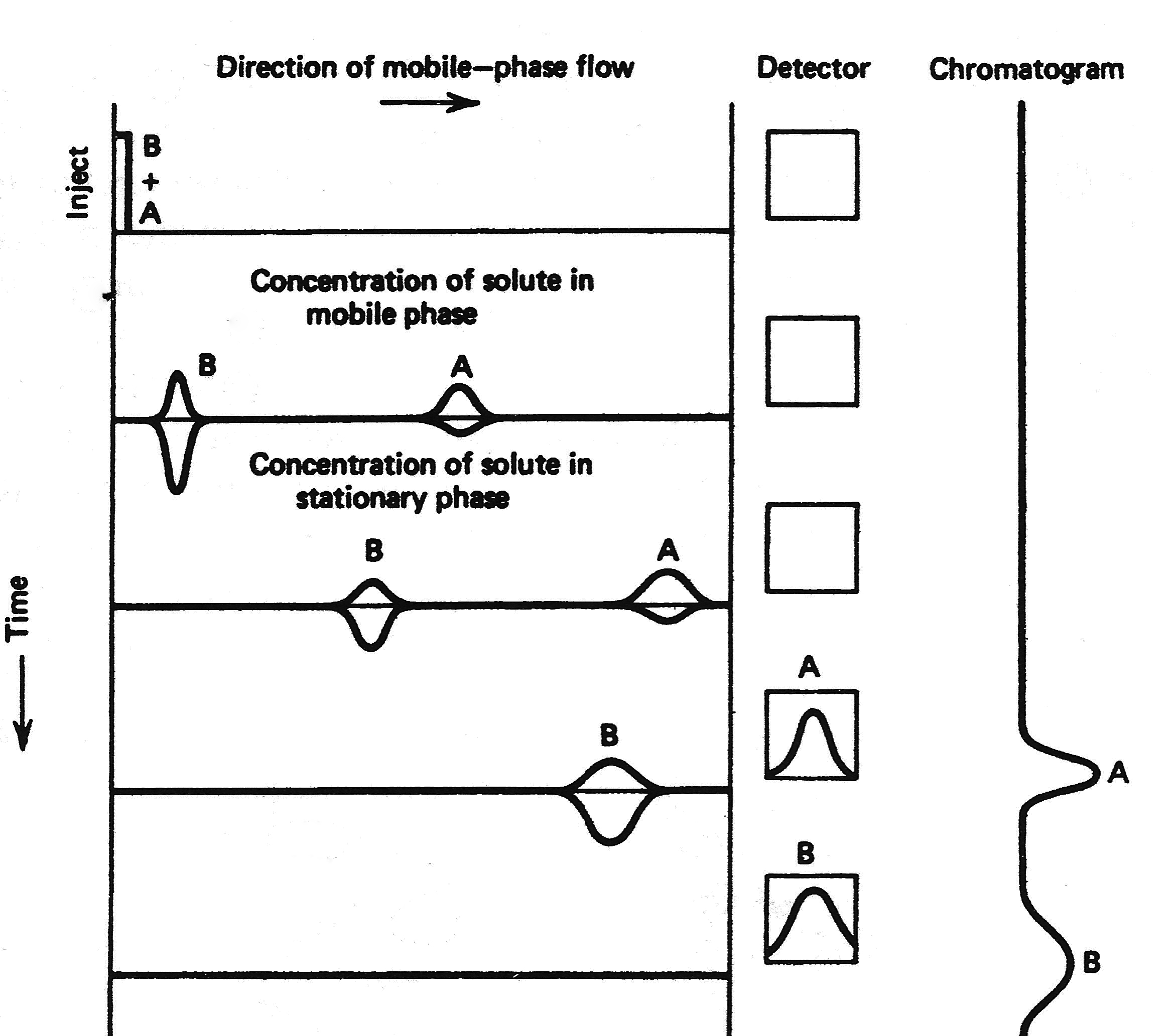
Durante una separación por CG, la muestra es vaporizada y transportada por la fase gaseosa móvil (es decir, el gas portador) a través de la columna. La separación de los diferentes componentes se logra en función de su presión relativa de vapor y afinidades para la fase estacionaria. La afinidad de una sustancia hacia la fase estacionaria puede describirse en términos químicos como una constante de equilibrio llamada constante de distribución Kc, también conocida como coeficiente de partición,\ ref {1}, donde [A] s es la concentración del compuesto A en la fase estacionaria y [A] m es la concentración del compuesto A en la fase móvil.
\[ K_{c} = [A]_{s}/[A]_{m} \label{1} \]
La constante de distribución (K c) controla el movimiento de los diferentes compuestos a través de la columna, por lo que las diferencias en la constante de distribución permiten la separación cromatográfica. La Figura\(\PageIndex{4}\) muestra una representación esquemática del proceso cromatográfico. K c depende de la temperatura, y también depende de la naturaleza química de la fase estacionaria. Así, la temperatura puede ser utilizada como una forma de mejorar la separación de diferentes compuestos a través de la columna, o una fase estacionaria diferente.
Un Cromatograma Típico
La figura\(\PageIndex{5}\) muestra un cromatograma del análisis de metanol residual en biodiesel, que es una de las propiedades requeridas que se deben medir para asegurar la calidad del producto en el momento y lugar de entrega.
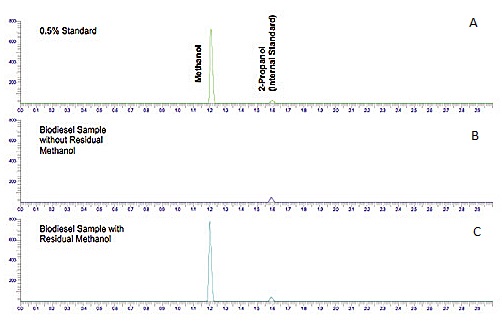
El cromatograma (Figura\(\PageIndex{5}\) a) muestra una solución estándar de metanol con 2-propanol como patrón interno. De la figura se puede observar que el metanol tiene una mayor afinidad por la fase móvil (menor K c) que el 2-propanol (iso-propanol), y por lo tanto eluye primero. Los cromatogramas (Figura\(\PageIndex{5}\) b y c) muestran dos muestras de biodiesel, una con metanol (Figura\(\PageIndex{5}\) b) y otra sin detección de metanol. Se agregó el estándar interno a ambas muestras con fines de cuantificación.
Descripción general del instrumento
Componentes de un sistema de cromatógrafo de gases
La Figura\(\PageIndex{6}\) muestra un diagrama esquemático de los componentes de un cromatógrafo de gases típico, mientras que la Figura\(\PageIndex{7}\) muestra una fotografía de un cromatógrafo de gases típico acoplado a un espectrómetro de masas (GC/MS).
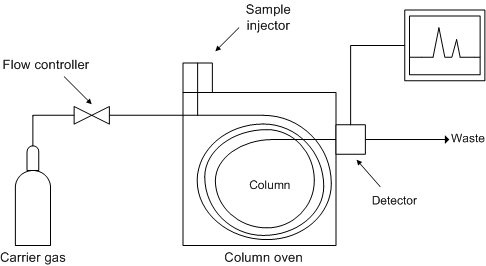

Gas portador
El papel del gas portador -fase móvil GC- es transportar las moléculas de muestra a lo largo de la columna mientras no se disuelven o adsorben en la fase estacionaria. El gas portador es inerte y no interactúa con la muestra, por lo que la selectividad de la separación de GC puede atribuirse a la fase estacionaria sola. Sin embargo, la elección del gas portador es importante para mantener una alta eficiencia. El efecto de diferentes gases portadores sobre la eficiencia de la columna está representado por la van Deemter (columnas empaquetadas) y la ecuación de Golay (columnas capilares). La ecuación de van Deemter,\ ref {2}, describe los tres efectos principales que contribuyen al ensanchamiento de banda en columnas empaquetadas y, como consecuencia, a una eficiencia reducida en el proceso de separación.
\[ HEPT\ =\ A+\frac{B}{u} + Cu \label{2} \]
Estos tres factores son:
- la difusión de remolino (el término A), que resulta del hecho de que en columnas empaquetadas los espacios entre las partículas a lo largo de la columna no son uniformes. Por lo tanto, algunas moléculas toman vías más largas que otras, y también hay variaciones en la velocidad de la fase móvil.
- la difusión molecular longitudinal (el término B) que es consecuencia de tener regiones con diferentes concentraciones de analito.
- la transferencia de masa en la fase líquida estacionaria (el término C)
El ensanchamiento se describe en términos de la altura equivalente a una placa teórica, HEPT, en función de la velocidad lineal promedio del gas, u. Un pequeño valor de HEPT indica un pico estrecho y una mayor eficiencia.
Dado que las columnas capilares no tienen ningún empaquetamiento, la ecuación de Golay,\ ref {3}, no tiene un término A. La ecuación de Golay tiene 2 términos C, uno para transferencia de masa en fase luego estacionaria (C s) y otro para transferencia de masa en la fase móvil (C M).
\[ HEPT\ =\ \frac{B}{u} \ +\ (C_{s}\ +\ C_{M})u \label{3} \]
El hidrógeno, el helio y el nitrógeno de alta pureza se utilizan comúnmente para la cromatografía de gases. Además, dependiendo del tipo de detector utilizado, se prefieren diferentes gases.
Inyector
Este es el lugar donde la muestra se volatiliza y se introduce cuantitativamente en la corriente de gas portador. Por lo general, se usa una jeringa para inyectar la muestra en el puerto de inyección. Las muestras se pueden inyectar manual o automáticamente con dispositivos mecánicos que a menudo se colocan encima del cromatógrafo de gases: los automuestreadores.
Columna
La columna cromatográfica de gases puede considerarse el corazón del sistema GC, donde se lleva a cabo la separación de los componentes de la muestra. Las columnas se clasifican como columnas empaquetadas o capilares. En la Tabla se muestra una comparación general de columnas empaquetadas y capilares\(\PageIndex{1}\). Las imágenes de columnas empaquetadas se muestran en la Figura\(\PageIndex{8}\) y Figura\(\PageIndex{9}\).
| Tipo de Columna | Columna Empacada | Columna Capilar |
|---|---|---|
| Historia | Primer tipo de columna GC utilizada | Tecnología moderna. Hoy en día la mayoría de las aplicaciones GC se desarrollan usando columnas capilares |
| Composición | Empaquetado con partículas de sílice sobre las que se recubre la fase estacionaria. | No embalado con material particulado. Hecho de sílice tratada químicamente cubierta con películas delgadas y uniformes en fase líquida. |
| Eficiencia | Bajo | Alto |
| Diámetro exterior | 2-4 mm | 0.4 mm |
| Longitud de columna | 2-4 metros | 15-60 metros |
| Ventajas | Muestras más grandes y de menor costo | Más rápido, mejor para mezclas complejas |


Dado que las aplicaciones más comunes empleadas hoy en día utilizan columnas capilares, nos centraremos en este tipo de columnas. Para definir una columna capilar, se deben especificar cuatro parámetros:
- La fase estacionaria es el parámetro que determinará la resolución final obtenida, e influirá en otros parámetros de selección. Cambiar la fase estacionaria es la forma más poderosa de alterar la selectividad en el análisis de GC.
- La longitud está relacionada con la eficiencia general de la columna y con el tiempo de análisis general. Una columna más larga aumentará la eficiencia máxima y la calidad de la separación, pero también aumentará el tiempo de análisis. Una de las compensaciones clásicas en las separaciones por cromatografía de gases (GC) se encuentra entre la velocidad de análisis y la resolución de picos.
- El diámetro interno de la columna (ID) puede influir en la eficiencia de la columna (y por lo tanto en la resolución) y también en la Al disminuir el diámetro interno de la columna, se pueden lograr mejores separaciones, pero la sobrecarga de la columna y el ensanchamiento de picos pueden convertirse en un problema.
- La capacidad de muestra de la columna también dependerá del grosor de la película. Además, la retención de los componentes de la muestra se verá afectada por el grosor de la película, y por lo tanto su tiempo de retención. Se puede lograr un tiempo de ejecución más corto y una mayor resolución usando películas delgadas, sin embargo estas películas ofrecen menor capacidad.
Detector
El detector detecta una propiedad fisicoquímica del analito y proporciona una respuesta que se amplifica y se convierte en una señal electrónica para producir un cromatograma. La mayoría de los detectores utilizados en GC fueron inventados específicamente para esta técnica, excepto el detector de conductividad térmica (TCD) y el espectrómetro de masas. En total, se han utilizado aproximadamente 60 detectores en GC. Los detectores que presentan una respuesta mejorada a ciertos tipos de analitos se conocen como “detectores selectivos”.
Durante los últimos 10 años ha habido un uso creciente de GC en combinación con espectrometría de masas (EM). El espectrómetro de masas se ha convertido en un detector estándar que permite menores límites de detección y no requiere la separación de todos los componentes presentes en la muestra. La espectroscopia de masas es uno de los tipos de detección que proporciona la mayor información con solo microgramos de muestra. La identificación cualitativa de compuestos desconocidos así como el análisis cuantitativo de muestras es posible mediante GC-MS. Cuando GC se acopla a un espectrómetro de masas, los compuestos que eluyen de la columna GC se ionizan mediante el uso de electrones (EI, ionización electrónica) o un reactivo químico (CI, ionización química). Los fragmentos cargados se enfocan y aceleran en un analizador de masas: normalmente un analizador de masas cuadrupolo. Los fragmentos con diferentes relaciones de masa a carga generarán diferentes señales, por lo que se detectará cualquier compuesto que produzca iones dentro del rango de masa del analizador de masas. Se pueden lograr límites de detección de 1-10 ng o incluso valores más bajos (por ejemplo, 10 pg) seleccionando el modo de escaneo apropiado.
Técnicas de preparación de muestras
Derivatización
La cromatografía de gases se utiliza principalmente para el análisis de compuestos volátiles térmicamente estables. Sin embargo, al tratar con muestras no volátiles, se pueden realizar reacciones químicas en la muestra para aumentar la volatilidad de los compuestos. Los compuestos que contienen grupos funcionales como OH, NH, CO 2 H y SH son difíciles de analizar por GC porque no son suficientemente volátiles, pueden ser atraídos demasiado fuertemente a la fase estacionaria o son térmicamente inestables. Las reacciones de derivatización más comunes utilizadas para GC se pueden dividir en tres tipos:
- Sililación.
- Acilación.
- Alquilación y Esterificación.
Las muestras se derivatizan antes de ser analizadas para:
- Aumentar la volatilidad y disminuir la polaridad del compuesto
- Reducir la degradación térmica
- Aumentar la sensibilidad incorporando grupos funcionales que conducen a señales de detector más altas
- Mejore la separación y reduzca la cola
Ventajas y desventajas
GC es la técnica analítica principal para la separación de compuestos volátiles. Varias características como velocidad de análisis, facilidad de operación, excelentes resultados cuantitativos y costos moderados habían ayudado a GC a convertirse en una de las técnicas más populares a nivel mundial.
Ventajas de GC
- Debido a su alta eficiencia, GC permite la separación de los componentes de mezclas complejas en un tiempo razonable.
- Cuantificación precisa (generalmente se obtienen picos reproducibles agudos)
- Técnica madura con muchas notas de aplicaciones disponibles para los usuarios.
- Están disponibles múltiples detectores con alta sensibilidad (ppb), los cuales también pueden ser utilizados en serie con un espectrómetro de masas ya que la EM es una técnica no destructiva.
Desventajas de GC
- Limitado a compuestos térmicamente estables y volátiles.
- La mayoría de los detectores GC son destructivos, excepto para MS.
Cromatografía de gases versus cromatografía líquida de alto rendimiento (HPLC)
A diferencia de la cromatografía de gases, que no es adecuada para moléculas no volátiles y térmicamente frágiles, la cromatografía líquida puede separar de manera segura una amplia gama de compuestos orgánicos, desde metabolitos de fármacos de moléculas pequeñas hasta péptidos y proteínas.
| GC | HPLC |
|---|---|
| La muestra debe ser volátil o derivatizada antes del análisis GC | La volatilidad no es importante, sin embargo la solubilidad en la fase móvil se vuelve crítica para el análisis. |
| La mayoría de los analitos tienen un peso molecular (MW) inferior a 500 Da (debido a problemas de volatilidad) | No hay límite de peso molecular superior en la medida en que la muestra pueda disolverse en la fase móvil apropiada |
| Se pueden acoplar a MS. Varias bibliotecas espectrales de masas están disponibles si se usa ionización electrónica (por ejemplo, http://chemdata.nist.gov/) | Los métodos deben adaptarse antes de usar un detector MS (no se pueden usar tampones no volátiles) |
| Se puede acoplar a varios detectores dependiendo de la aplicación | Para algunos detectores el solvente debe ser un problema. Al cambiar los detectores, algunos métodos requerirán una modificación previa |