
4.11: Espectrometría de Masas
- Page ID
- 71103

Principios de Espectrometría de Masas y Aplicaciones Modernas
La espectrometría de masas (EM) es una poderosa técnica de caracterización utilizada para la identificación de una amplia variedad de compuestos químicos. En su forma más simple, la EM es simplemente una herramienta para determinar el peso molecular de las especies químicas en una muestra. Sin embargo, con la alta resolución que se puede obtener de las máquinas modernas, es posible distinguir isómeros, isótopos e incluso compuestos con pesos moleculares nominalmente idénticos. Se han compilado bibliotecas de espectros de masas que permiten una rápida identificación de la mayoría de los compuestos conocidos, incluyendo proteínas de hasta 100 kDa (100,000 amu).
Los espectrómetros de masas separan los compuestos en función de una propiedad conocida como la relación masa/carga. La muestra a identificar primero se ioniza, y luego se pasa a través de alguna forma de campo magnético. Con base en parámetros como el tiempo que tarda la molécula en recorrer una cierta distancia o la cantidad de deflexión causada por el campo, se puede calcular una masa para el ion. Como se discutirá más adelante, existe una amplia variedad de técnicas para ionizar y detectar compuestos.
Las limitaciones de la EM generalmente provienen de compuestos que no son fácilmente ionizables, o que se descomponen tras la ionización. Los isómeros geométricos generalmente se pueden distinguir fácilmente, pero las diferencias en quiralidad no se resuelven fácilmente. Las complicaciones también pueden surgir de muestras que no se disuelven fácilmente en solventes comunes.
Técnicas de ionización
Impacto de electrones (EI)
En la ionización por impacto de electones, una muestra vaporizada se hace pasar a través de un haz de electrones. El haz de alta energía (típicamente 70 eV) elimina los electrones de las moléculas de muestra dejando una especie radical cargada positivamente. El ion molecular es típicamente inestable y experimenta descomposición o reordenamiento para producir iones fragmentos. Debido a esto, el impacto electrónico se clasifica como una técnica de ionización “dura”. Con respecto a los compuestos que contienen metales, los fragmentos en EI casi siempre contendrán el átomo metálico (es decir, [ML n] +• fragmentos a [ML n-1] + L •, no ML n-1 • + L +). Una de las principales limitaciones de la IE es que la muestra debe ser volátil y térmicamente estable.
Ionización química (CI)
En la ionización química, la muestra se introduce en una cámara llena de exceso de gas reactivo (como metano). El gas reactivo es ionizado por electrones, formando un plasma con especies como CH 5 +, que reaccionan con la muestra para formar el ion pseudomolecular [M+H] +. Debido a que el IC no implica reacciones radicales, la fragmentación de la muestra es generalmente mucho menor que la de la IE. El CI también se puede operar en modo negativo (para generar aniones) mediante el uso de diferentes gases reactivos. Por ejemplo, una mezcla de CH 4 y NO 2 generará iones hidróxido, que pueden abstraer protones para producir la especie [M-H] -. Una técnica relacionada, la ionización química a presión atmosférica (APCI) entrega la muestra como una pulverización neutra, la cual luego se ioniza por descarga en corona, produciendo iones de manera similar a la descrita anteriormente. El APCI es particularmente adecuado para especies no polares de bajo peso molecular que no pueden ser fácilmente analizadas por otras técnicas comunes como la ESI.
Ionización/desorción de campo
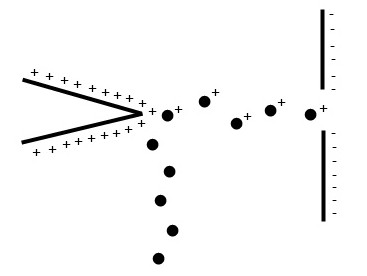
La ionización y desorción de campo son dos técnicas estrechamente relacionadas que utilizan tunelización cuántica de electrones para generar iones. Típicamente, se aplica un potencial altamente positivo a un electrodo con una punta afilada, dando como resultado un alto gradiente de potencial en la punta Figura\(\PageIndex{1}\). A medida que la muestra alcanza este campo, se produce un túnel de electrones para generar el catión, el cual es repelido en el analizador de masas. La ionización de campo utiliza muestras gaseosas mientras que en la desorción de campo la muestra se adsorbe directamente sobre el electrodo. Ambas técnicas son blandas, dando como resultado iones de baja energía que no se fragmentan fácilmente.
Ionización por Electrospray (ESI)
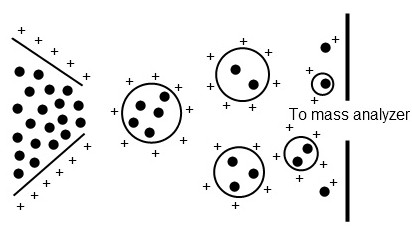
En ESI, se genera un aerosol altamente cargado a partir de una muestra en solución. A medida que las gotitas se contraen debido a la evaporación, la densidad de carga aumenta hasta que se produce una explosión culómbica, produciendo gotitas hijas que repiten el proceso hasta que se generan iones de muestra individualizados (Figura\(\PageIndex{2}\). Una de las limitaciones de es el requisito de que la muestra sea soluble. ESI se aplica mejor a compuestos cargados, polares o básicos.
Ionización por desorción láser asistida por matriz (MALDI)
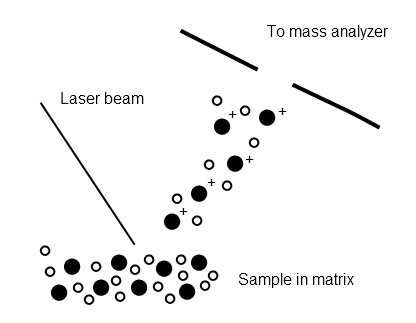
La ionización por desorción láser genera iones por ablación de una superficie usando un láser pulsado. Esta técnica se mejora enormemente mediante la adición de una matriz co-cristalizada con la muestra. A medida que se irradia la muestra, se genera un penacho de moléculas desorbidas. Se cree que la ionización ocurre en este penacho debido a una variedad de interacciones químicas y físicas entre la muestra y la matriz (Figura\(\PageIndex{3}\)). Una de las principales ventajas de MALDI es que produce iones de carga individual casi exclusivamente y se puede utilizar para volatilizar especies de peso molecular extremadamente alto como polímeros y proteínas. Una técnica relacionada, la ionización por desorción sobre silicio (DIOS) también utiliza desorción láser, pero la muestra se inmoviliza sobre una superficie porosa de silicio sin matriz. Esto permite el estudio de compuestos de bajo peso molecular que pueden ser oscurecidos por picos de matriz en MALDI convencional.
Espectrometría de masas de plasma acoplado inductivamente (ICP-MS)
Se utiliza un soplete de plasma generado por inducción electromagnética para ionizar muestras. Debido a que la temperatura efectiva del plasma es de aproximadamente 10,000 °C, las muestras se descomponen en iones de sus elementos constituyentes. Así, se pierde toda la información química y la técnica es la más adecuada para el análisis elemental. ICP-MS se utiliza típicamente para el análisis de oligoelementos.
Bombardeo rápido de átomos (FAB) y espectrometría de masas de iones secundarios (SIMS)
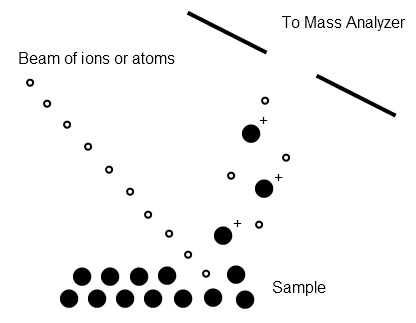
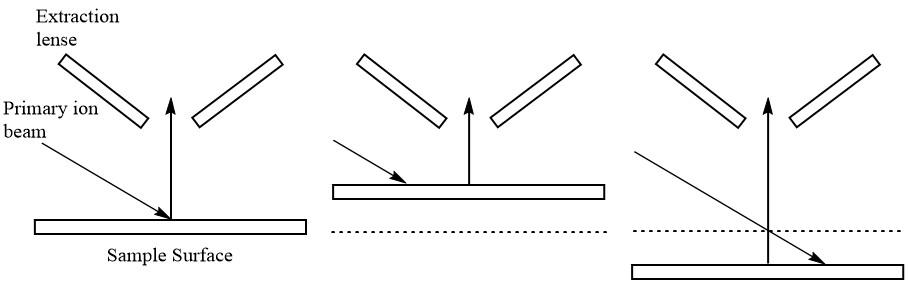
Ambas técnicas implican la pulverización catódica de una muestra para generar iones individualizados; FAB utiliza una corriente de átomos de gas inerte (argón o xenón) mientras que SIMS utiliza iones como Cs +. La ionización ocurre por transferencia de carga entre los iones y la muestra o por protonación del material de la matriz (Figura\(\PageIndex{4}\)). Se pueden analizar tanto muestras sólidas como líquidas. Un aspecto único de estas técnicas para el análisis de sólidos es la capacidad de hacer perfiles de profundidad debido a la naturaleza destructiva de la técnica de ionización.
Elección de una técnica de ionización
Dependiendo de la información deseada del análisis de espectrometría de masas, se pueden desear diferentes técnicas de ionización. Por ejemplo, se puede usar un método de ionización dura como el impacto de electrones para una molécula compleja con el fin de determinar las partes componentes por fragmentación. Por otro lado, una muestra de alto peso molecular de polímero o proteína puede requerir un método de ionización como MALDI para ser volatilizada. A menudo, las muestras se pueden analizar fácilmente usando múltiples métodos de ionización, y la elección se simplifica para elegir el método más conveniente. Por ejemplo, la ionización por electronebulización puede acoplarse fácilmente a sistemas de cromatografía líquida, ya que no se requiere preparación adicional de muestras. \(\PageIndex{1}\)La tabla proporciona una guía rápida de las técnicas de ionización típicamente aplicadas a varios tipos de muestras.
| Información deseada | Técnica de ionización |
| Análisis elemental | Plasma acoplado inductivamente |
| Perfilado de profundidad | Bombardeo de átomos rápidos/espectroscopía de masas de iones secundarios |
| Análisis de especiación/componentes químicos (fragmentación deseada) | Impacto de electrones |
| Identificación de especies moleculares de compuestos solubles en solventes comunes | Ionización por electrospray |
| Identificación de especies moleculares de compuestos de hidrocarburos | Ionización de campo |
| Identificación de especies moleculares de compuestos de alto peso molecular | Ionización por desorción láser asistida por matriz |
| Identificación de especies moleculares de compuestos que contienen halógenos | Ionización química (modo negativo) |
Analizadores de Masa
Sectores
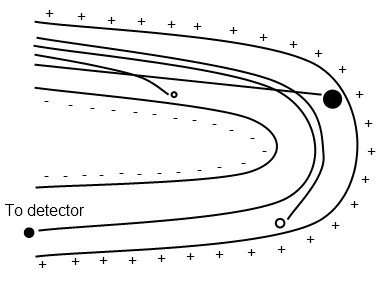
Se utiliza un campo magnético o eléctrico para desviar iones hacia trayectorias curvas dependiendo de la relación m/z, con iones más pesados que experimentan menos deflexión (Figura\(\PageIndex{5}\)). Los iones se ponen en foco en la hendidura del detector variando la intensidad del campo; se genera un espectro de masas explorando las intensidades de campo lineal o exponencialmente. Los analizadores de masas sectoriales tienen alta resolución y sensibilidad, y pueden detectar rangos de masa altos, pero son caros, requieren grandes cantidades de espacio, y son incompatibles con las técnicas de ionización más populares MALDI y ESI.
Tiempo de vuelo (TOF)
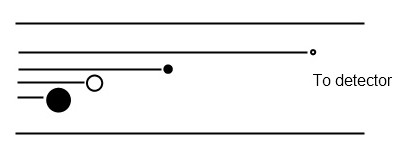
Se mide la cantidad de tiempo requerido para que un ion recorra una distancia conocida (Figura\(\PageIndex{6}\)). Un pulso de iones se acelera a través de un analizador eléctrico de tal manera que tienen energías cinéticas idénticas. Como resultado, su velocidad depende directamente de su masa. Se requieren condiciones de vacío extremadamente altas para extender la trayectoria libre media de los iones y evitar colisiones. Los analizadores de masas TOF son los más rápidos, tienen rangos de masa ilimitados y permiten la detección simultánea de todas las especies, pero se combinan mejor con fuentes de ionización pulsadas como MALDI.
Cuatripolo
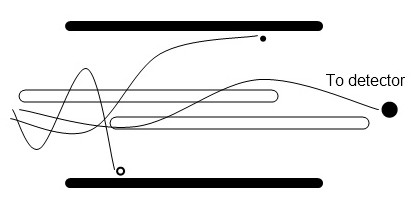
Los iones se pasan a través de cuatro varillas paralelas que aplican un voltaje variable y potencial de radiofrecuencia (Figura\(\PageIndex{7}\)). A medida que cambia el campo, los iones responden experimentando trayectorias complejas. Dependiendo del voltaje aplicado y las frecuencias de RF, solo los iones de cierta relación m/z tendrán trayectorias estables y pasarán por el analizador. Todos los demás iones se perderán por colisión con las varillas. Los analizadores cuadrupolares son relativamente económicos, pero tienen una resolución limitada y un rango de masa bajo.
Trampa de iones
Las trampas de iones operan bajo el mismo principio que el cuadrupolo, pero contienen los iones en el espacio. Los electrodos pueden ser manipulados para expulsar selectivamente iones de relaciones m/ z deseadas, permitiendo el análisis de masa. Las trampas de iones son especialmente adecuadas para ciclos repetidos de espectrometría de masas debido a su capacidad para retener iones de relaciones m/ z deseadas. Los fragmentos seleccionados pueden fragmentarse adicionalmente mediante disociación inducida por colisión con gas helio. Las trampas de iones son compactas, relativamente económicas y se pueden adaptar a muchos instrumentos híbridos.
Acoplamiento de espectrometría de masas a otros instrumentos
La espectrometría de masas es una herramienta poderosa para la identificación de compuestos, y frecuentemente se combina con técnicas de separación como la cromatografía líquida o gaseosa para la identificación rápida de los compuestos dentro de una mezcla. Por lo general, los sistemas de cromatografía líquida se emparejan con espectrómetros de masas ESI-cuadrupolo para aprovechar la muestra solvatada. Los sistemas GC-MS suelen emplear ionización por impacto electrónico y analizadores de masas cuadrupolo o trampa de iones para aprovechar las moléculas en fase gaseosa y las bibliotecas de fragmentación asociadas con EI para una identificación rápida.
Los espectrómetros de masas también se acoplan a menudo en tándem para formar sistemas MS-MS. Normalmente, el primer espectrómetro utiliza una técnica de ionización dura para fragmentar la muestra. Los fragmentos se pasan a un segundo analizador de masas donde pueden fragmentarse y analizarse adicionalmente. Esta técnica es particularmente importante para estudiar moléculas grandes y complejas como las proteínas.
Bombardeo de átomo rápido
El bombardeo rápido de átomos (FAB) es una técnica de ionización para espectroscopía de masas empleando espectroscopía de masas de iones secundarios (SIMS). Antes de la aparición de esta técnica, solo había una forma limitada de obtener el espectro de masas del oligopéptido intacto que no es fácil de vaporizar. Antes de 1970, la ionización electrónica (EI) o la ionización química (IC) era ampliamente utilizada pero esos métodos requieren la vaporización destructiva de la muestra. La desorción de campo de iones con fisión nuclear superó este problema, aunque debido a la necesidad de una técnica especial y la fisión nuclear de 252 Cf limita la generalidad de este enfoque. FAB se hizo prevalente resolviendo los problemas subyacentes mediante el bombardeo de átomos rápidos o iones que tienen alta energía cinética sobre la muestra en la matriz.
Principio
El FAB utiliza el bombardeo de haces acelerados de átomos o iones y la muestra ionizada es emitida por la colisión de los haces y la muestra en matriz. En esta sección se discute el detalle de cada paso.
Haz Átomo
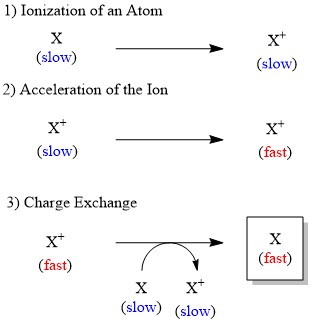
Aunque los iones pueden ser acelerados por el campo eléctrico con relativa facilidad, ese no es el caso del átomo neutro. Por lo tanto, en la FAB la conversión de átomo neutro en ion es significativa para generar las especies aceleradas. El átomo rápido como el xenón utilizado para el bombardeo se produce a través de tres etapas (Figura\(\PageIndex{8}\)):
- Ionización del átomo por colisión con electrón.
- Aceleración del ion generado a través de alto potencial eléctrico.
- Transferencia de electrones del ion acelerado a otro átomo lento, proporcionando el átomo acelerado deseado.
Haz de iones
De la misma manera que el haz atómico, también se puede utilizar un haz de iones rápido. Aunque a menudo se emplea el ion cesio (Cs +) más barato y más pesado que el xenón, tienen el inconveniente de que la espectroscopia de masas puede estar contaminada por los iones.
Bombardeo
El átomo o ion rápido obtenido es luego bombardeado a la muestra en matriz que es un tipo de disolvente que tiene alto punto de ebullición, resultando en transferencia de momento y vaporización de la muestra (Figura\(\PageIndex{9}\)). El átomo rápido utilizado para el bombardeo se denomina haz primario de átomos o iones mientras que el haz secundario de átomos o iones corresponde a los iones pulverizados y neutrales. La muestra ionizada es dirigida por la óptica de iones, lo que lleva a la detección de esos iones en el analizador de masas.
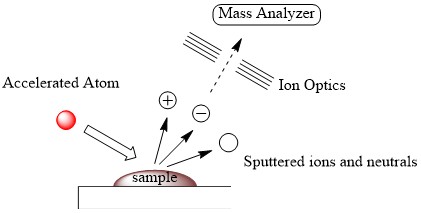
Matrices
Una de las características cruciales de FAB es el uso de matriz líquida. Por ejemplo, la señal de larga duración en FAB es responsable de usar la matriz. Debido a la condición de alto vacío, el disolvente habitual para laboratorio de química como el agua y otro disolvente orgánico común se excluye para FAB y, por lo tanto, es necesario emplear disolvente con alto punto de ebullición llamado matriz. En la tabla se\(\PageIndex{1}\) muestran ejemplos de matriz.
| Matrix | Iones observados (m/z) |
| glicerol | 93 |
| Tioglicerol | 109 |
| Alcohol 3-nitrobencílico (3-NOBA) | 154 |
| N-octil-3-nitrofenileter (NOP) | 252 |
| Trietanolamina | 150 |
| Dietanolamina | 106 |
| Polietilenglicol (mezclas) | Depende del glicol utilizado |
Instrumento
En la Figura se muestra una imagen de un instrumento típico para espectrometría de masas de bombardeo atómico rápido\(\PageIndex{10}\).

Spectra
El espectro obtenido por FAB tiene información de estructura o naturaleza de enlace del compuesto además de la masa. Aquí se muestran tres espectros como ejemplos.
glicerol
El espectro de masas FAB típico de glicerol solo se muestra en la Figura\(\PageIndex{11}\).
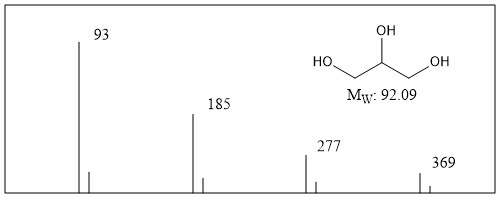
El glicerol muestra señal a m/z 93 que corresponde al glicerol protonado con satélite pequeño derivado del isótopo de carbono (13 C). Al mismo tiempo, las señales para racimo de glicerol protonado también se observan a menudo a m/z 185, 277 y 369. Como se ve en este ejemplo, también se puede detectar señal de agregación de la muestra y esto proporcionará la información de la muestra.
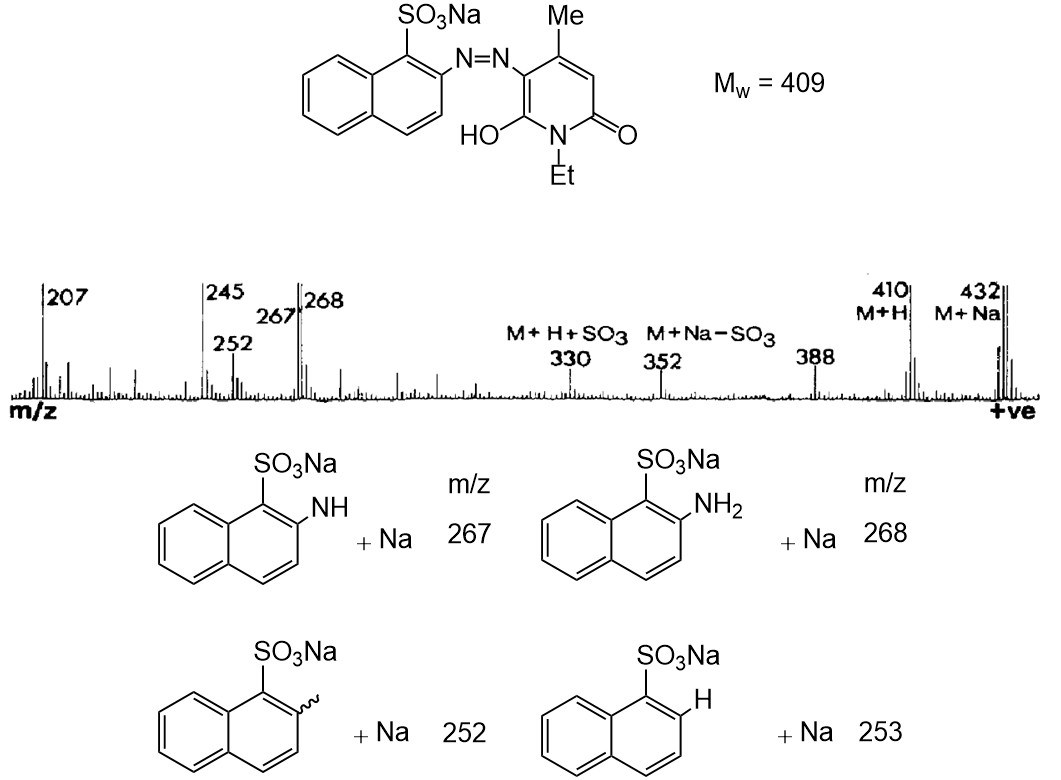
Compuesto Azo sulfonado
La Figura\(\PageIndex{12}\) muestra el espectro FAB positivo del compuesto azoico sulfonado X y la estructura de los fragmentos plausibles en el espectro. La señal del compuesto diana X (M w = 409) se observó a m/z 432 y 410 como aducto con sodio y protón, respectivamente. Debido a la presencia de algún tipo de enlaces relativamente débiles, se observaron varias fragmentaciones. Por ejemplo, la señal a m/z 352 y 330 resultó de la escisión del enlace aril-sulfonato. También se produjo la escisión del enlace nitrógeno-nitrógeno en el resto azo, produciendo la señal del fragmento a m/z 267 y 268. Además, tomando en cuenta que la formación favorable del triple enlace nitrógeno-nitrógeno a partir del resto azo, se puede escindir el enlace aril-nitrógeno y de hecho se detectaron a m/z 253 y 252. Como se muestra en estos ejemplos, la fragmentación puede ser utilizada para obtener información sobre la estructura y la naturaleza de enlace del compuesto deseado.
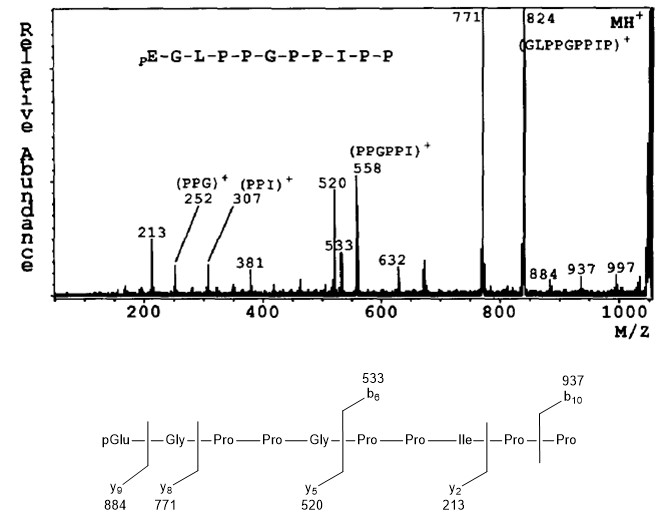
Potenciador de bradiquinina C
En la Figura se muestra el espectro de masas de la molécula protonada (MH + = m/z 1052) del potenciador C de bradiquinina\(\PageIndex{13}\). En este caso se produce fragmentación entre ciertos aminoácidos, proporcionando la información de la secuencia peptídica. Por ejemplo, la señal a m/z 884 corresponde al fragmento como resultado de la escision del enlace Gly-Leu. Cabe señalar que el patrón de fragmentación no solo se realiza por un tipo de escisión de enlaces. La fragmentación en el enlace entre Gly-Pro es un buen ejemplo; se observan dos tipos de fragmentos (m/z 533 y 520). Así, el patrón de fragmentación puede proporcionar la información de la secuencia del péptido.
Espectrometría de masas de iones secundarios (SIMS)
La espectrometría de masas de iones secundarios (SIMS) es un método analítico que tiene límites de detección muy bajos, es capaz de analizar en un amplio rango dinámico, tiene alta sensibilidad y alta resolución de masa. En esta técnica, los iones primarios se utilizan para pulverizar una superficie sólida (y a veces líquida) de cualquier composición. Esto provoca la emisión de electrones, iones y especies neutras, llamadas partículas secundarias, desde la superficie sólida. Luego se analizan los iones secundarios mediante un espectrómetro de masas. Dependiendo del modo de funcionamiento seleccionado, los SIMS se pueden utilizar para la composición de la superficie y el análisis de la estructura química, el perfilado de profundidad y las imágenes.
Teoría
De todas las partículas secundarias que se pulverizan desde la superficie de la muestra, solo aproximadamente 1 de cada 1,000 se emite como un ion. Debido a que solo los iones pueden ser detectados por espectrometría de masas, es importante comprender cómo se forman estos iones secundarios.
Modelos de pulverización catódica
La pulverización catódica se puede definir como la emisión de átomos, moléculas o iones desde una superficie objetivo como resultado del bombardeo de partículas de la superficie. Este fenómeno ha sido descrito por dos conjuntos diferentes de modelos.
El primer enfoque para describir la pulverización catódica, llamada teoría de cascada de colisión lineal, compara los átomos con las bolas de billar y asume que las colisiones atómicas son completamente elásticas. Aunque hay algunos tipos diferentes de pulverización catódica definidos por este modelo, el tipo que es más importante para SIMS es la pulverización catódica por colisión lenta. En este tipo de pulverización catódica, el ion primario choca con la superficie del objetivo y provoca una cascada de colisiones aleatorias entre los átomos en el objetivo. Eventualmente, estas colisiones aleatorias resultan en la emisión de un átomo desde la superficie objetivo, como puede verse en la Figura\(\PageIndex{14}\). Este modelo no toma en cuenta la ubicación de los átomos- solo requiere que la energía del ion entrante sea mayor que la energía requerida para sublimar los átomos de la superficie objetivo.
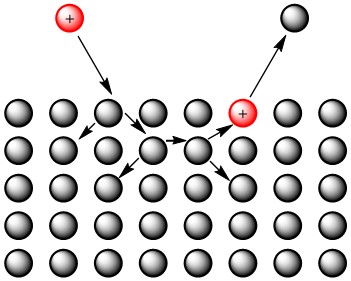
A pesar de que este método realiza simplificaciones excesivas con respecto a las interacciones atómicas y la estructura, sus datos de rendimiento de pulverización catódica pronosticados están bastante cerca de los datos experimentales para elementos como Cu, Zn, Ag y Au, que tienen altos rendimientos de pulverización catódica. Sin embargo, para elementos de bajo rendimiento por pulverización catódica, el modelo predice tres veces más iones pulverizados de lo que realmente se observa.
El segundo método para describir la pulverización catódica utiliza modelos tridimensionales generados por computadora de los átomos y moléculas en la muestra para predecir el efecto del bombardeo de partículas. Todos los modelos bajo esta categoría describen el sólido diana en términos de sus átomos y moléculas constituyentes y sus interacciones entre sí. Sin embargo, estos modelos solo toman en cuenta las fuerzas atómicas (no las fuerzas electrónicas) y describen el comportamiento atómico utilizando la mecánica clásica (no la mecánica cuántica). Dos ejemplos específicos de este tipo de modelos son:
- El modelo de dinámica molecular
- La aproximación binaria de colisión.
Modelos de ionización
Los modelos de ionización de la pulverización catódica se pueden dividir en dos categorías, teorías que predicen la ionización fuera del objetivo y teorías que predicen que se generan dentro del objetivo. En las teorías que describen la ionización fuera del objetivo, la partícula primaria golpea el objetivo, provocando la emisión de un átomo o molécula excitada del objetivo. Esta partícula se relaja emitiendo un electrón Auger, convirtiéndose así en un ion. Debido a que no se ha descrito ninguna ecuación matemática simple para esta teoría, es de poco uso práctico. Por esta razón, la ionización dentro de los modelos objetivo se utiliza con mayor frecuencia. Adicionalmente, se ha demostrado que la ionización ocurre con mayor frecuencia dentro del objetivo. Aunque hay muchos modelos que describen la ionización dentro del objetivo, dos modelos representativos de este tipo son el modelo de ruptura de enlaces y la teoría del equilibrio térmico local.
En el modelo de ruptura de enlaces, la partícula primaria golpea la diana y provoca la escisión heterolítica de un enlace en la diana. Entonces, ya sea un anión o un catión se emite directamente desde la superficie objetivo. Este es un modelo importante a mencionar porque tiene implicaciones útiles. Dicho simplemente, el rendimiento de iones positivos puede ser incrementado por la presencia de átomos electronegativos en el objetivo, en el haz de iones primarios, o en la cámara de muestra en general. Lo contrario también es cierto: el rendimiento de iones negativos puede aumentarse por la presencia de átomos electropositivos.
La teoría del equilibrio térmico local puede describirse como una expansión del modelo de ruptura de enlaces. Aquí, se dice que el aumento en el rendimiento de iones positivos cuando el objetivo está en presencia de átomos electronegativos es el resultado de la barrera de alto potencial del óxido metálico que se forma. Esto da como resultado una baja probabilidad de que el ion secundario sea neutralizado por un electrón, dando así un alto rendimiento de iones positivos.
Instrumentación
Fuentes de iones primarios
Los iones primarios en un instrumento SIMS (etiquetado como “Fuente de iones primarios” en la Figura\(\PageIndex{15}\)) son generados por uno de los tres tipos de pistolas de iones. El primer tipo, llamado fuente de plasma de bombardeo de electrones, utiliza electrones acelerantes (producidos a partir de un filamento calentado) para bombardear un ánodo. Si la energía de estos electrones es de dos a tres veces mayor que la energía de ionización del átomo, se produce la ionización. Una vez que se obtiene un cierto número de iones y electrones, se forma un plasma. Después, se utiliza un extractor para hacer un haz de iones enfocado a partir del plasma.
En el segundo tipo de fuente, llamada fuente de metal líquido, una película de metal líquido fluye sobre una aguja roma. Cuando esta película es sometida a un fuerte campo eléctrico, los electrones son expulsados de los átomos en el metal líquido, dejándolos ionizados. Luego, un extractor dirige los iones fuera de la pistola de iones.
La última fuente se llama fuente de ionización superficial. Aquí, los átomos de baja energía de ionización son absorbidos en un metal de alta función de trabajo. Este tipo de sistema permite la transferencia de electrones desde los átomos superficiales al metal. Cuando se incrementa la temperatura, salen más átomos (o iones) de la superficie que los que absorben en la superficie, provocando un aumento en los iones absorbidos en comparación con los átomos absorbidos. Eventualmente, casi todos los átomos que salen de la superficie están ionizados y pueden ser utilizados como haz de iones.
El tipo de fuente utilizada depende del tipo de experimento SIMS que se va a ejecutar así como la composición de la muestra a analizar. Una comparación de las tres fuentes diferentes se da en la Tabla\(\PageIndex{2}\).
| Fuente | Tamaño de punto (µm) | Brillo (A/m 2 Sr) | Velocidad de energía (eV) | Tipo Ion |
| Plasma de Bombardeo de Electrones | 1 | 10 4 -10 7 | <10 | Ar +, Xe +, O 2 + |
| Metal Líquido | 0.05 | 10 10 | >10 | Ga +, In +, Cs + |
| Ionización superficial | 0.1 | 10 7 | <1 | Cs + |
De las tres fuentes, el plasma de bombardeo de electrones tiene el mayor tamaño de punto. Por lo tanto, esta fuente tiene un haz de alto diámetro y no tiene la mejor resolución espacial. Por esta razón, esta fuente se utiliza comúnmente para el análisis masivo como el perfilado de profundidad. La fuente de metal líquido es ventajosa para la formación de imágenes SIMS porque tiene una alta resolución espacial (o tamaño de punto bajo). Por último, la fuente de ionización superficial funciona bien para SIMS dinámicos (ver arriba)
porque su dispersión de energía muy pequeña permite una tasa de grabado uniforme.
Además del tipo de pistola de iones, también es importante la identidad del ion primario. O 2 + y Cs + se utilizan comúnmente porque mejoran el rendimiento de iones secundarios positivos o negativos, respectivamente. Sin embargo, el uso de la fuente de plasma de gas inerte es ventajoso porque permite estudios de superficie sin reaccionar con la propia superficie. El uso de la fuente de plasma O 2 + permite una mayor producción de iones secundarios cargados positivamente, pero alterará la superficie que se está estudiando. Además, un ion primario pesado permite una mejor resolución de profundidad porque no penetra tan lejos en la muestra como un ion ligero.
Sputtering
La velocidad de pulverización catódica, o el número de iones secundarios que se eliminan de la superficie de la muestra por bombardeo por un ion primario, depende tanto de las propiedades del objetivo como de los parámetros del haz primario.
Hay muchos factores objetivo que afectan la tasa de pulverización catódica. Algunos ejemplos son la estructura cristalina y la topografía del objetivo. Específicamente, los cristales hexagonales empaquetados y las superficies rugosas dan el mayor rendimiento de pulverización catódica. Hay muchas otras propiedades del blanco que efectúan la pulverización catódica, pero no se discutirán aquí.
Como se discutió anteriormente, diferentes fuentes de iones primarios se utilizan para diferentes aplicaciones SIMS. Además de la fuente utilizada, también es importante la manera en que se utiliza la fuente. Primero, la tasa de pulverización catódica se puede aumentar aumentando la energía del haz. Por ejemplo, el uso de un haz de energía superior a 10 keV da un máximo de 10 partículas de pulverización catódica por impacto de iones primarios. En segundo lugar, aumentar la masa de iones primarios también aumentará el rendimiento de iones secundarios. Por último, el ángulo de incidencia también es importante. Se ha encontrado que se puede lograr una tasa máxima de pulverización catódica si el ángulo de impacto es de 70° con respecto a la superficie normal.
Espectrómetros de Masas
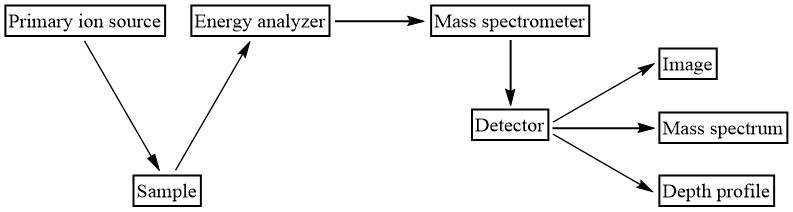
El detector que mide la cantidad y tipo de iones secundarios pulverizados desde la superficie de la muestra es un espectrómetro de masas. Consulte la Figura\(\PageIndex{15}\) para ver un diagrama que muestra dónde está el espectrómetro de masas en relación con los demás componentes del instrumento. El tipo de análisis que se desea hacer determina qué tipo de espectrómetro se utiliza. Tanto los SIMS dinámicos como los estáticos suelen utilizar un analizador de masa de sector magnético porque tiene una alta resolución de masa. Los SIMS estáticos (así como los SIMS de imágenes) también pueden usar un sistema de tiempo de vuelo, que permite una alta transmisión. Una descripción de cómo funciona cada uno de estos espectrómetros de masas y cómo se detectan los iones se puede encontrar en otra parte (ver https://cnx.org/contents/kl4gTdhf@1/Principles-of-Mass-Spectrometry-and-Modern-Applications).
Muestras
Los SIMS pueden ser utilizados para analizar la superficie y aproximadamente 30 µm por debajo de la superficie de casi cualquier muestra sólida y algunas muestras líquidas. Dependiendo del tipo de análisis SIMS elegido, es posible obtener datos tanto cualitativos como cuantitativos sobre la muestra.
Selección de Técnica
Hay tres tipos principales de experimentos SIMS: SIMS dinámicos, SIMS estáticos y SIMS de imágenes.
En el análisis SIMS dinámico, el objetivo se pulveriza a una tasa alta. Esto permite el análisis a granel cuando el espectrómetro de masas se escanea en todos los rangos de masa para obtener un espectro de masas y se toman múltiples mediciones en diferentes áreas de la muestra. Si el espectrómetro de masas está configurado para analizar rápidamente las masas individuales secuencialmente a medida que el objetivo se erosiona rápidamente, es posible ver la profundidad a la que se ubican átomos específicos hasta 30 µm por debajo de la superficie de la muestra. Este tipo de análisis se denomina perfil de profundidad. El perfilado de profundidad es muy útil porque es un método cuantitativo, permite el cálculo de la concentración en función de la profundidad siempre y cuando se utilicen estándares implantados de iones y se mida la profundidad del cráter. Consulte la sección anterior para obtener más información sobre implantes iónicos.
Los SIMS también se pueden usar para obtener una imagen de una manera similar a la SEM mientras se da una mejor sensibilidad que el SEM. Aquí, un haz de iones finamente enfocado (en lugar de un haz de electrones, como en SEM) se escanea por ráster sobre la superficie objetivo y los iones secundarios resultantes se analizan en cada punto. Usando la identidad de los iones en cada punto analizado, se puede ensamblar una imagen basada en las distribuciones de estos iones.
En SIMS estáticos, la superficie de la muestra se erosiona muy lentamente de manera que los iones que se emiten provienen de áreas que aún no han sido alteradas por el ion primario. Al hacer esto, es posible identificar los átomos y algunas de las moléculas justo en la superficie de la muestra.
Un ejemplo que muestra la utilidad de SIMS es el análisis de huellas dactilares utilizando este instrumento. Se han empleado muchas otras formas de análisis para caracterizar la composición química de las huellas dactilares como la GC-MS. Esto es importante en la ciencia forense para determinar la degradación de huellas dactilares, para detectar explosivos o narcóticos, y para ayudar a determinar la edad de la persona que dejó la huella mediante el análisis de diferencias en las secreciones sebáceas. Comparado con GC-MS, SIMS es una mejor opción de análisis porque es relativamente menos destructivo. Para poder realizar una GC-MS, la huella dactilar debe ser disuelta. SIMS, por otro lado, es un método de estado sólido. Además, debido a que SIMS solo se erosiona a través de unas pocas monocapas, la huella dactilar puede conservarse para futuros análisis y para mantener registros. Adicionalmente, el perfil de profundidad SIMS permite al investigador determinar el orden en que se tocaron las sustancias. Por último, se puede obtener una imagen de la huella digital utilizando el análisis SIMS de imagen.
Preparación de Muestras
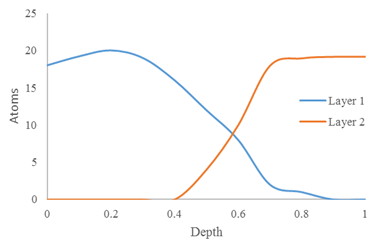
Al igual que con cualquier otro análisis instrumental, SIMS requiere alguna preparación de muestras. Primero, las muestras rugosas pueden requerir pulido debido a que la textura desigual se mantendrá a medida que la superficie sea rociada. Debido a que los átomos superficiales son el analito en las imágenes y los SIMS estáticos, obviamente no se requiere pulido. Sin embargo, se requiere para el perfilado de profundidad. Sin pulir, las capas debajo de la superficie de la muestra aparecerán mezcladas con la capa superior en el espectro, como se puede ver en la Figura\(\PageIndex{16}\).
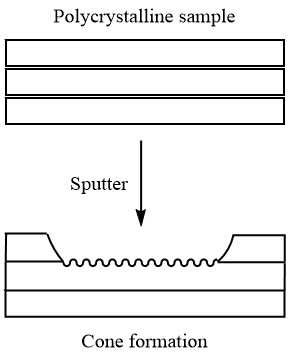
Pero, el pulido antes del análisis no necesariamente garantiza incluso la pulverización catódica. Esto se debe a que las diferentes orientaciones de los cristales se rocían a diferentes velocidades. Entonces, si la muestra es policristalina o tiene límites de grano (esto suele ser un problema con las muestras metálicas), la muestra puede desarrollar pequeños conos donde se está produciendo la pulverización catódica, lo que lleva a un perfil de profundidad inexacto, como se ve en la Figura\(\PageIndex{17}\).
El análisis de aisladores usando SIMS también requiere una preparación especial de la muestra como resultado de la acumulación de carga eléctrica en la superficie (ya que el aislante no tiene una trayectoria conductora para difundir la carga). Esto es un problema porque distorsiona los espectros observados. Para evitar la carga superficial, es una práctica común recubrir la muestra con una capa conductora como el oro.
Una vez preparada la muestra para su análisis, debe montarse en el portamuestras. Existen algunos métodos para hacer esto. Una forma es colocar la muestra en un portamuestras cargado por resorte que empuja la muestra contra una máscara. Este método es ventajoso porque el investigador no tiene que preocuparse por ajustar la altura de la muestra para diferentes muestras (vea a continuación para averiguar por qué la altura de la muestra es importante). Sin embargo, debido a que la máscara está encima de la muestra, es posible bombardear accidentalmente la máscara. Otro método utilizado para montar muestras es simplemente pegarlas a una placa de respaldo usando epoxi plateado. Este método requiere secarlo bajo una lámpara de calor para asegurar que todos los volátiles se evaporen del pegamento antes del análisis. Alternativamente, la muestra se puede prensar en un metal blando como el indio. Los dos últimos métodos son especialmente útiles para el montaje de muestras aislantes, ya que proporcionan una trayectoria conductora para ayudar a prevenir la acumulación de carga.
Al cargar la muestra montada en el instrumento, es importante que la altura de la muestra con respecto a la lente del instrumento sea correcta. Si la muestra está demasiado cerca o demasiado lejos, los iones secundarios o bien no se detectarán o se detectarán en el borde del cráter siendo producidos por los iones primarios (ver Figura\(\PageIndex{18}\)). Idealmente, los iones secundarios que se analizan deben ser los resultantes del centro del haz primario donde la energía e intensidad son más uniformes.
Estándares
Para hacer análisis cuantitativos usando SIMS, es necesario utilizar estándares de calibración ya que la tasa de ionización depende tanto del átomo (o molécula) como de la matriz. Estos estándares suelen estar en forma de implantes iónicos que pueden depositarse en la muestra usando un implantador o usando el haz de iones primario del SIMS (si la fuente de iones primarios es filtrada en masa). Al comparar la concentración conocida de iones implantados con el número de iones de implante pulverizados, es posible calcular el valor del factor de sensibilidad relativa (RSF) para el ión de implante en la muestra particular. Al comparar este valor de RSF con el valor en una tabla RSF estándar y ajustar todos los valores de RSF de la tabla por la diferencia entre ellos, es posible calcular las concentraciones de otros átomos en la muestra. Para obtener más información sobre los valores de RSF, ver arriba.
A la hora de elegir un isótopo para su implantación iónica, es importante tener en cuenta las posibles interferencias de masa. Por ejemplo, 11 B, 16 O y 27 Al tienen las mismas masas globales e interferirán entre sí con la intensidad de iones en los espectros. Por lo tanto, se debe elegir un implante iónico que no tenga la misma masa que cualquier otra especie en la muestra que sea de interés.
También es importante la profundidad a la que se deposita el implante. El ion implantado debe ser inferior a la profundidad de equilibrio, por encima de la cual, se produce la pulverización catódica caótica hasta alcanzar un equilibrio de pulverización catódica. Sin embargo, se debe tener cuidado para que los iones implantados no pasen por la capa de interés en la muestra; si la matriz cambia, los iones implantados ya no se bombardearán a la misma velocidad, provocando que las concentraciones sean inexactas.
Efectos Matrix
En SIMS, los efectos de la matriz son comunes y se originan a partir de cambios en la eficiencia de ionización (el número de especies ionizadas en comparación con el número total de especies pulverizadas) y el rendimiento de pulverización catódica. Una de las principales causas de los efectos matriciales es el haz primario. Como se discutió anteriormente, los iones primarios electronegativos aumentan el número de iones secundarios cargados positivamente, mientras que los iones primarios electropositivos aumentan el número de iones secundarios cargados negativamente. Los efectos de la matriz también pueden ser causados por especies presentes en la muestra. Las consecuencias de estos efectos matriciales dependen de la identidad de las especies efectoras y de la composición de la muestra. Para corregir los efectos matriciales, es necesario utilizar un estándar y comparar los resultados con RSF (ver arriba).
Límites de detección
Para la mayoría de los átomos, los SIMS pueden detectar con precisión hasta una concentración de 1 ppm. Para algunos átomos, se puede lograr una concentración de 10 ppb. El límite de detección en este instrumento se establece por la tasa de conteo (cuántos iones se pueden contar por segundo) en lugar de por una limitación debida a la masa del ion. Entonces, para disminuir el límite de detección, la muestra puede ser bombardeada a una tasa mayor.
Sensibilidad
La sensibilidad del análisis SIMS depende del elemento de interés, la matriz en la que se encuentra el elemento y qué ion primario se utiliza. La sensibilidad de SIMS hacia un ion particular se puede determinar fácilmente mirando una tabla de RSF. Entonces, por ejemplo, al mirar una tabla de RSF para un ion primario de oxígeno y iones secundarios positivos, se muestra que los metales alcalinos tienen la mayor sensibilidad (tienen valores bajos de RSF). Esto tiene sentido, ya que estos átomos tienen las afinidades electrónicas más bajas y son los más fáciles de ionizar. De igual manera, al observar la tabla RSF para un haz de iones primarios de cesio e iones secundarios negativos, se observa que los halógenos tienen la mayor sensibilidad. Nuevamente, esto tiene sentido ya que los halógenos tienen las mayores afinidades electrónicas y aceptan electrones fácilmente.
Interpretación de datos
Se pueden obtener tres tipos de espectros a partir de un análisis SIMS. A partir de SIMS estáticos, se produce un espectro de masas. A partir de SIMS dinámicos, se produce un perfil de profundidad o espectro de masas. Y, como era de esperar, se produce una imagen a partir de SIMS de imagen.
Espectros de Masa
Al igual que con un espectro de masas típico, la relación masa/carga (m/z) se compara con la intensidad de iones. Sin embargo, debido a que SIMS es capaz de un rango dinámico de 9 órdenes de magnitud, la intensidad de los espectros de masas SIMS se muestra en una escala logarítmica. A partir de estos datos, es posible observar datos isotópicos así como datos de iones moleculares y sus abundancias relativas en la superficie de la muestra.
Perfil de Profundidad
Un perfil de profundidad muestra la intensidad de uno o más iones con respecto a la profundidad (o, equivalentemente, tiempo). Se debe tener precaución al interpretar estos datos- si los iones se recogen de la pared del cráter en lugar de desde la parte inferior, parecerá que la capa en cuestión corre más profunda en la muestra de lo que realmente lo hace.
Ionización por desorción láser asistida por matriz (MALDI)
Desarrollo de MALDI
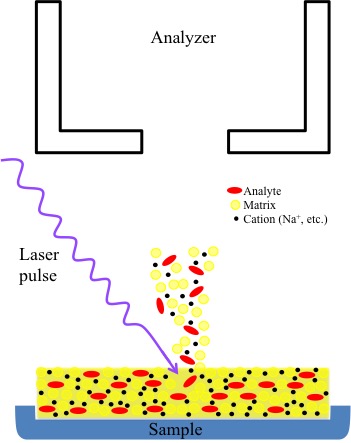
Como se aludió en secciones anteriores, la desorción láser (LD) se desarrolló originalmente para producir iones en fase gaseosa. Esto se logra mediante la pulsación de un láser sobre la superficie de la muestra para ablacionar el material causando la ionización y vaporización de las partículas de la muestra. Sin embargo, la probabilidad de alcanzar un espectro de masas valioso depende en gran medida de las propiedades del analito. Además, las masas observadas en el espectro fueron productos de la fragmentación molecular si el peso molecular estaba por encima de 500 Da. Claramente, esta no fue una instrumentación óptima para analizar biomoléculas grandes y compuestos bioinorgánicos que no se ionizan bien y las muestras se degradaron durante el proceso. La ionización por desorción láser asistida por matriz (MALDI) se desarrolló y alivió muchos problemas asociados con las técnicas de LD. La técnica MALDI permite detectar proteínas con masas de hasta 300,000 Da. Esto es importante para la química bioinorgánica cuando se visualizan productos resultantes de reacciones catalíticas, modificaciones de metaloenzimas y otras aplicaciones.
MALDI como proceso disminuye la cantidad de daño a la muestra al proteger los analitos individuales dentro de una matriz (más información de las matrices más adelante). La propia matriz absorbe gran parte de la energía introducida por el láser durante la acción pulsante. Además, la energía absorbida por la matriz se transfiere posteriormente al analito (Figura\(\PageIndex{19}\)). Una vez energizado, el analito se ioniza y se libera en un penacho de iones que contienen cationes comunes (Na +, K +, etc.), iones de matriz e iones analito. Estos iones luego ingresan al tubo de vuelo donde son enviados al detector. Diferentes modos instrumentales se ajustan para las diferencias en el tiempo de vuelo de iones (Figura\(\PageIndex{19}\)). La técnica MALDI también es más sensible y universal ya que los reajustes para igualar la frecuencia de absorción no son necesarios debido a la absorción de la matriz. Muchas de las matrices de uso común tienen absorciones de longitud de onda similares Tabla\(\PageIndex{3}\).
| Matrix | Longitud de onda | Aplicación | Estructura |
| Ácido ciano-4-hidroxicinámico | UV: 337 nm, 353 nm | Péptidos | 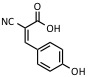 |
| 6-aza-2-tiotimina | UV: 337 nm, 353 nm | Proteínas, péptidos, complejos no covalentes | 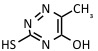 |
| k, m, N-di (tri) hidroxi-acetofenona |
UV: 337 nm, 353 nm |
Proteínas, péptidos, complejos no covalentes | 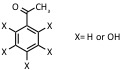 |
| Ácido 2,5-dihidroxibenzoico (requiere 10% de ácido 2-hidroxi-5-metoxibenzoico) | UV: 337 nm, 353 nm | Proteínas, péptidos, carbohidratos, polímeros sintéticos | 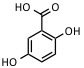 |
| Ácido sinapínico | UV: 337 nm, 353 nm | Proteínas, péptidos | 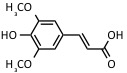 |
| Ácido nicotínico | UV: 266 nm | Proteínas, péptidos, formación de aductos | 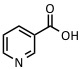 |
| Ácido succínico | IR: 2.94 µm, 2.79 µm | Proteínas, péptidos | 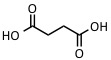 |
| glicerol | IR: 2.94 µm, 2.79 µm | Proteínas, péptidos | 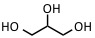 |
Colección de MALDI Spectra
El proceso de MALDI se lleva a cabo en 2 pasos:
- Preparación de muestras.
- Ablación de muestras
Preparación de Muestras
La muestra para análisis se combina con una matriz (un disolvente que contiene pequeñas moléculas orgánicas que tienen una fuerte absorbancia a la longitud de onda del láser) y se agrega a la placa MALDI (Figura\(\PageIndex{18}\)). Después, la muestra se seca a la superficie de la placa antes de analizarla, dando como resultado la matriz dopada con el analito de interés como una “solución sólida”. La Figura\(\PageIndex{20}\) muestra la carga de un péptido en agua en matriz de ácido ciano-4-hidroxicinámico.

Antes de la inserción de la placa en el instrumento MALDI, las muestras deben estar completamente secas. La placa MALDI con las muestras secas se coloca sobre un portador y se inserta en la cámara de vacío (Figura\(\PageIndex{21}\) a-b). Después de evacuar la cámara, está lista para iniciar el paso de ablación de la muestra.

Después de cargar la muestra en el instrumento, la cámara del instrumento mostrará activar para mostrar una transmisión en vivo desde el interior de la cámara. La transmisión en vivo permite al controlador ver la ubicación donde se está adquiriendo el espectro. Esto se vuelve especialmente importante cuando el operador dispara manualmente los pulsos láser.
Colección de un Espectro
Cuando la muestra se carga en la cámara de vacío del instrumento, existen varias opciones para tomar un espectro de masas. Primero, existen varios modos para el instrumento, dos de los cuales se describen aquí: modos axial y reflectron.
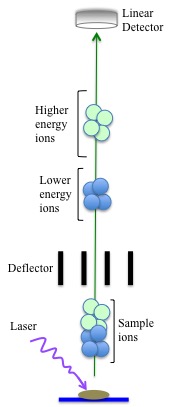
Modo Axial
En el modo axial (o lineal), solo se requiere un pulso de iones muy corto antes de que los iones bajen por el tubo de vuelo y golpeen el detector. Este modo se usa a menudo cuando no se requiere precisión exacta ya que la precisión de masa tiene un error de aproximadamente +/- 2-5%. Las fuentes de estos errores se encuentran en el tiempo de llegada de diferentes iones a través del tubo de vuelo al detector. Los errores en el tiempo de llegada son causados por la diferencia en la velocidad inicial con la que viajan los iones en función de su tamaño. Los iones más grandes tienen una menor velocidad inicial, por lo que llegan al detector después de un periodo de tiempo más largo. Esto disminuye la resolución de detección de masa.
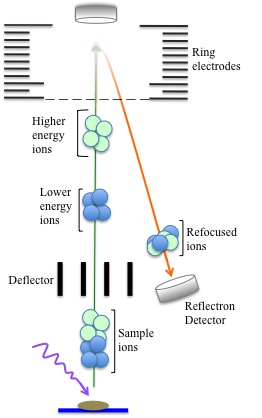
Modo Reflectron
En el modo reflectron (“espejo iónico”), los iones se reenfocan antes de que golpeen el detector. El reflectron en sí es en realidad un conjunto de electrodos de anillo que crean un campo eléctrico que es constante cerca del extremo del tubo de vuelo. Esto hace que los iones se ralenticen e inviertan la dirección hacia un detector separado. Luego, los iones más pequeños se acercan a los iones grandes antes de que el grupo de iones golpee el detector. Esto ayuda a mejorar la resolución de detección y disminuye el error de precisión a +/- 0.5%.
Ejemplo de aplicación MALDI
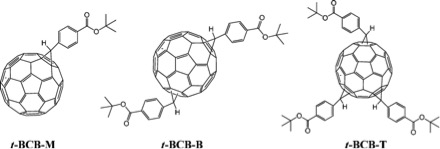
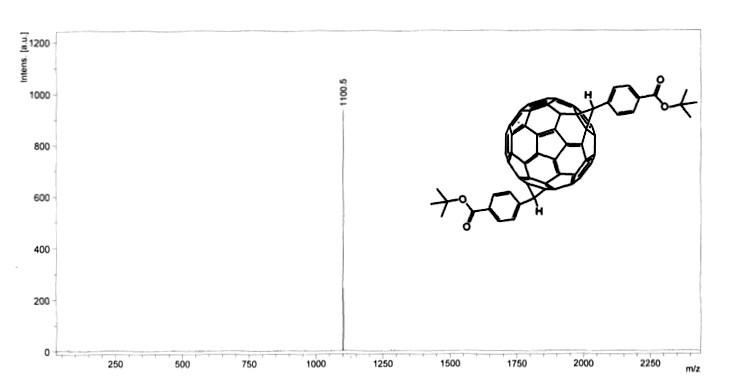
Si bien MALDI se usa ampliamente en el análisis de proteínas y péptidos, también se usa para analizar nanomateriales. El siguiente ejemplo describe el análisis de análogos de fullereno sintetizados para un sistema de conversión de alto rendimiento para energía solar. El fullereno C 60 es una molécula esférica de carbono compuesta por 60 sp 2 átomos de carbono, cuyas propiedades pueden alterarse a través de la funcionalización. Se sintetizaron y aislaron una serie de fullerenos funcionalizados con ferc-butil-4-C61-benzoato (t-BCb). El MALDI no se utilizó extensivamente como método para observar la actividad, sino que se utilizó como técnica conformativa para determinar la presencia del producto deseado. Se sintetizaron tres derivados de fullereno (Figura\(\PageIndex{24}\)) .La identidad y número de grupos funcionales se determinaron usando MALDI (Figura\(\PageIndex{25}\)).
Espectrometría de masas de desorción/ionización láser asistida por superficie (SALDI-MS)
La espectrometría de masas de desorción/ionización láser asistida por superficie, conocida como SALDI-MS, es una técnica de espectrometría de masas blandas capaz de analizar todo tipo de moléculas orgánicas pequeñas, polímeros y biomoléculas grandes. El principio esencial de este método es similar al de (espectrometría de masas de desorción/ionización láser asistida por matriz) MALDI-MS (ver http://cnx.org/contents/925e204d-d85...3e4d60057b37@1), pero la matriz orgánica comúnmente utilizada en MALDI se ha cambiado a la superficie de ciertos sustratos, generalmente compuestos inorgánicos. Esto convierte a SALDI en una técnica de ionización libre de matriz que evita la interferencia de las moléculas de la matriz.
Se considera que el SALDI es un proceso de tres etapas que se muestra en la Figura\(\PageIndex{26}\).
- Las muestras se mezclan con los sustratos que proporcionan una gran área de superficie para soportar la molécula de muestra.
- Las muestras son irradiadas con IR de pulsos láser UV cuando la energía de los pulsos láser es absorbida por los sustratos y transferida a las moléculas de muestra.
- Se inicia el proceso de desorción e ionización, lo que produce iones que se aceleran en el analizador.
Dado que la mayor parte del aporte de energía va a sustratos en lugar de moléculas de muestra, se cree que es una técnica de ionización suave útil en los campos de la química y la biología química.
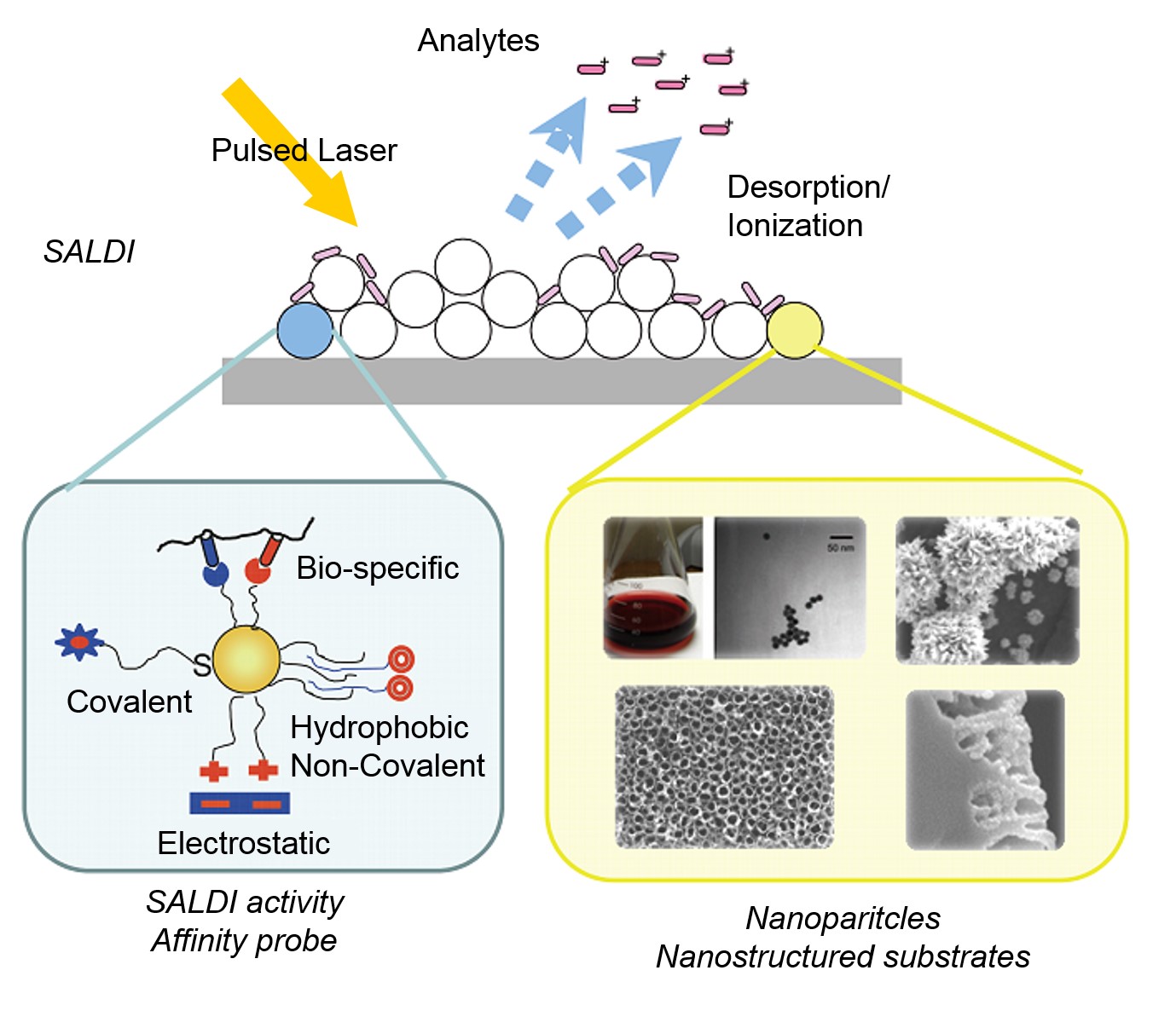
La característica más importante del sustrato en SALDI es una gran superficie. En los últimos 30 años, se han realizado esfuerzos para explorar nuevos materiales de sustrato que aumenten la sensibilidad y selectividad en SALDI-MS. Dependiendo de los compuestos sustrato que se utilicen, la interacción entre los materiales del sustrato y las moléculas de muestra podría ser covalente, no covalente como el efecto hidrófobo, bioespecífica como el reconocimiento entre biotinas y avidinas, y entre antígenos y anticuerpos, o electrostática. Con las características únicas señaladas anteriormente, SALDI es capaz de combinar las ventajas de las técnicas de ionización tanto duras como blandas. Por un lado, las moléculas de bajo peso molecular (LMW) podrían analizarse e identificarse en SALDI-MS, lo que se asemeja a la función de la mayoría de las técnicas de ionización dura. Por otro lado, los iones moleculares o cuasimoleculares dominarían los espectros como lo que comúnmente vemos en los espectros preparados por técnicas de ionización suave.
Historia
La técnica SALDI en realidad surgió de su conocida técnica rival, MALDI. El desarrollo de técnicas de ionización blanda, que incluyeron principalmente MALDI y ESI, permitió a químicos y biólogos químicos analizar grandes polímeros y biomoléculas mediante espectrometría de masas. Esto debe atribuirse al proceso de ionización suave que prohibió un gran grado de fragmentación que complicó los espectros, y los iones resultantes fueron predominantemente iones moleculares o iones cuasimoleculares. En otras palabras, la tolerancia a las impurezas se incrementaría ya que los espectros se simplificaron altamente. Si bien fue efectivo para determinar el peso molecular de los analitos, los picos de la matriz también aparecerían en un rango de masa baja, lo que interfería seriamente con el análisis de analitos de LMW. Como resultado, surgió el método SALDI para resolver el problema reemplazando la matriz por una superficie que era más bien estacionaria.
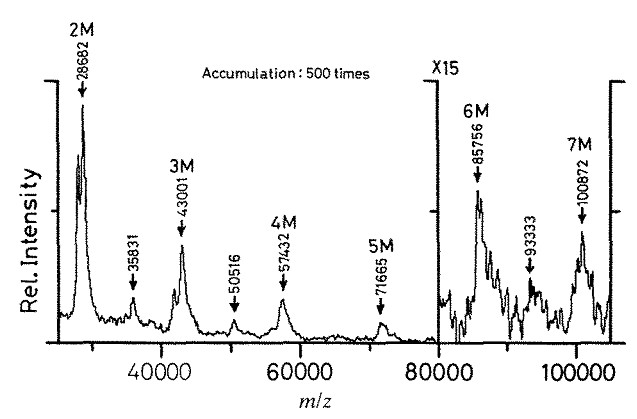
La idea original de SALDI fue planteada por Tanaka (Figura\(\PageIndex{27}\)) en 1988. Los polvos de cobalto ultrafinos con un diámetro promedio de aproximadamente 300 Å que se mezclaron en la muestra fueron responsables de “calentamiento rápido” debido a su alta fotoabsorción y baja capacidad calorífica. Con una gran superficie, los polvos de cobalto fueron capaces de conducir el calor a grandes cantidades de glicerol líquido circundante y moléculas de analito, lo que de hecho resultó en un mecanismo de desorción/ionización térmica. El límite de masa superior se incrementó hasta 100 kDa, lo que se muestra en la Figura\(\PageIndex{28}\) para el análisis de lisozimas de clara de huevo de gallina.

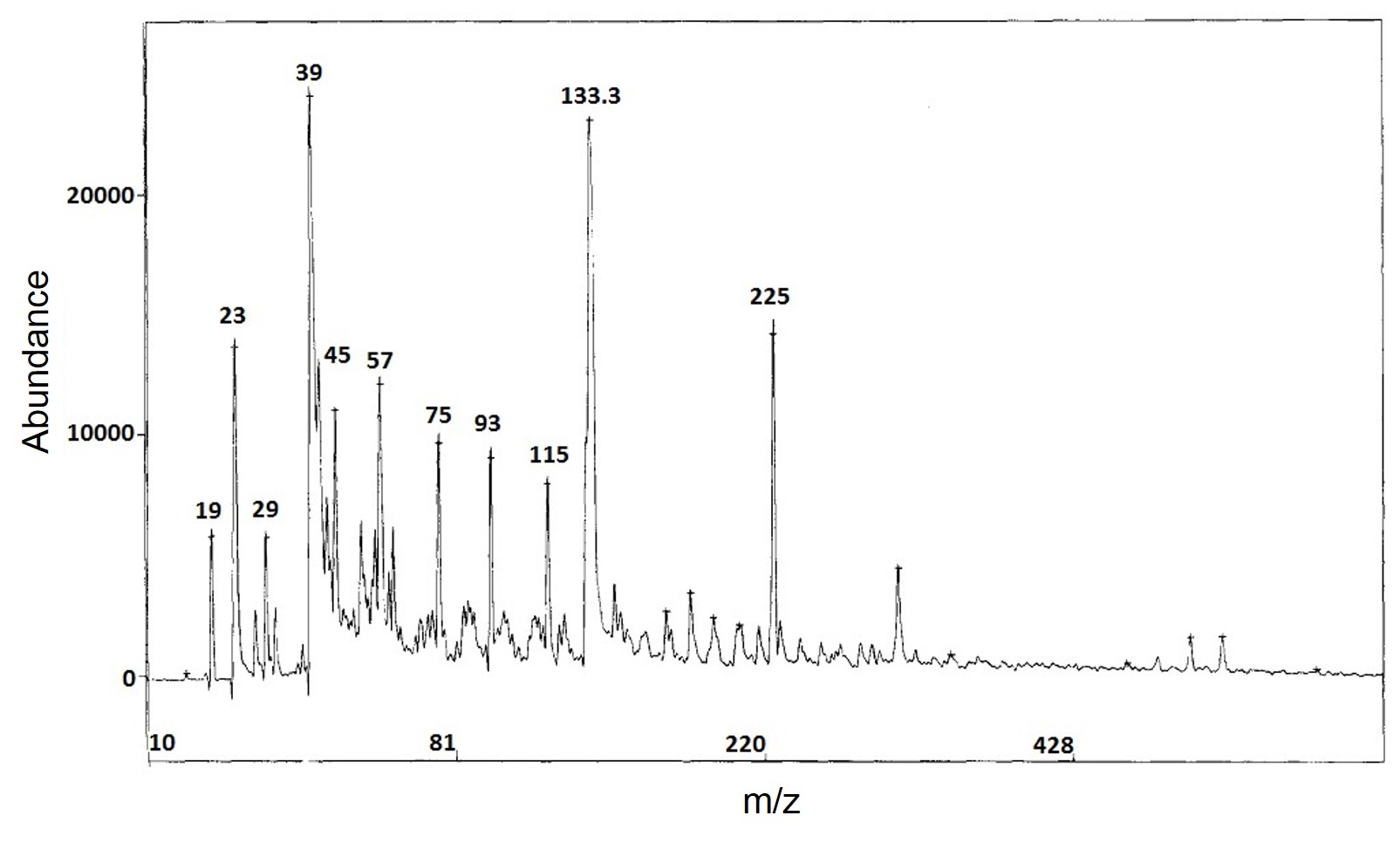
Al principio no se prestó mucha atención al rango de masa baja, y el concepto de “superficie asistida” no se propuso hasta que Sunner (Figura\(\PageIndex{29}\)) y sus compañeros de trabajo reportaron el estudio sobre la SALDI de grafito en 1995. Y esa fue la primera vez que los químicos usaban el término “SALDI”. Lograron obtener espectros de masas tanto de proteínas como de analitos LWM irradiando mezcla de partículas de grafito de 2-150 μm y soluciones de analitos en glicerol. Aunque la fragmentación de las moléculas de glicerol LMW fue relativamente complicada (Figura\(\PageIndex{30}\)), aún se consideró como una mejora significativa en la ionización de moléculas pequeñas por métodos de ionización blanda.

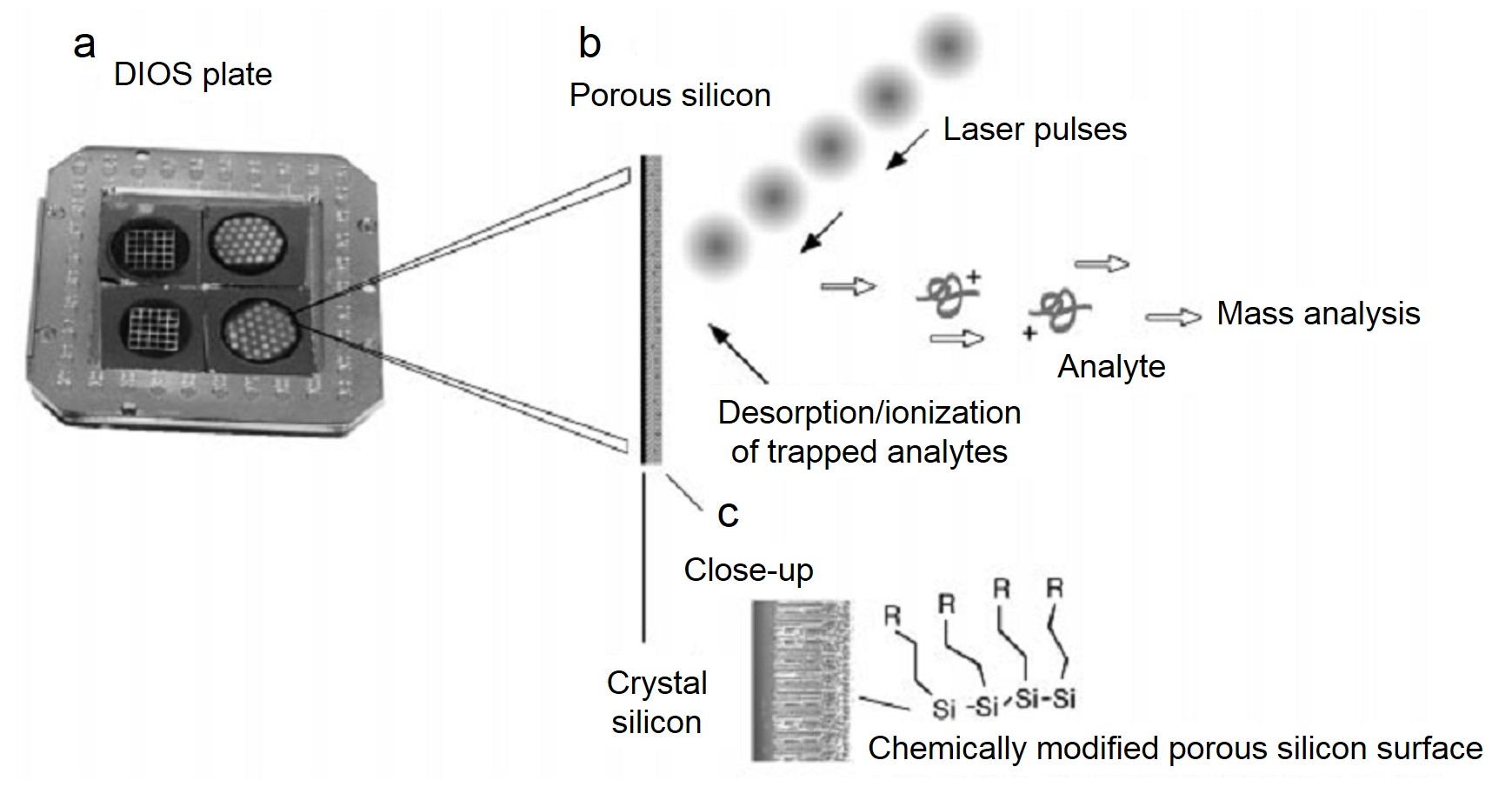
A pesar del avance mencionado anteriormente, la SALDI no interesó ampliamente a los químicos. Independientemente de sus inconvenientes en el límite de masa superior para el análisis de moléculas grandes, la sensibilidad estuvo lejos de ser satisfactoria en comparación con las técnicas de ionización dura en términos de pruebas de moléculas LMW. Esta situación ha cambiado desde que se introdujeron los nanomateriales como sustratos, especialmente para el desarrollo exitoso de la desorción/ionización sobre silicio poroso (DIOS) que se muestra en la Figura\(\PageIndex{31}\). De hecho, la mayoría de las investigaciones sobre SALDI-MS se han centrado en la explotación de nuevos sustratos nanomateriales, con el objetivo de ampliar aún más el rango de masas, mejorar la reproducibilidad, potenciar la sensibilidad y extender las categorías de compuestos que pudieron ser analizados. Hasta el momento, se han utilizado una variedad de nanomateriales en SALDI-MS, incluyendo nanomateriales basados en carbono, nanomateriales a base de metales, nanomateriales basados en semiconductores, etc.
Principios Básicos de SALDI
Mecanismo de Desorción e Ionización
Como técnica de ionización suave, se espera que SALDI produzca iones moleculares o cuasimoleculares en los espectros de masas finales. Dado que esto requiere que el proceso de ionización sea efectivo y controlable, lo que significa que suficientes moléculas de muestra podrían ionizarse mientras que la mayor parte de la fragmentación debe evitarse.
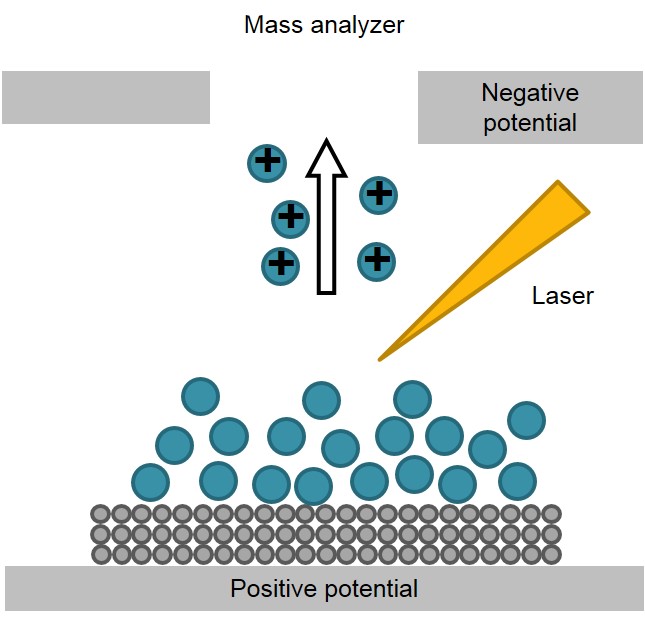
Si bien el objetivo original mencionado anteriormente se ha logrado con éxito durante años, el estudio sobre el mecanismo de desorción e ionización en detalle sigue siendo una de las áreas de investigación más populares y polémicas de la SALDI en la actualidad. Se concuerda principalmente en que el material del sustrato ha jugado un papel importante tanto en la activación como en la protección de las moléculas de analito. La imagen esquemática que describe todo el proceso se muestra en la Figura\(\PageIndex{33}\). La entrada de energía del láser pulsado es absorbida en gran medida por el material del sustrato, lo que posiblemente es seguido por una complicada transferencia de energía desde el material del sustrato a las moléculas de analito absorbidas. Como resultado, se pudo desencadenar tanto la desorción térmica como la no térmica, y para diferentes modos de experimentos de SALDI, el proceso específico de desorción e ionización difiere mucho.
El mecanismo para la superficie porosa de silicio como sustrato de SALDI ha sido ampliamente estudiado por investigadores. En general, el proceso se puede subdividir en los siguientes pasos:
- La adsorción de moléculas de analito neutras tiene lugar mediante la formación de enlaces de hidrógeno con grupos silanol superficiales;
- La excitación electrónica del sustrato bajo la influencia del pulso láser genera un par electrónico libre/ “agujero”. Esta separación provoca el enriquecimiento de cargas positivas cerca de la capa superficial; como resultado, la acidez de los grupos silanol aumenta y la transferencia de protones a analitos se vuelve más fácil;
- Los iones analito se activan térmicamente y, por lo tanto, se disocian de la superficie.
Cuando no hay ningún donante de protones asociado en las proximidades de moléculas de analito, la desorción podría ocurrir sin ionización. Posteriormente, la molécula de analito desorbida es ionizada en fase gaseosa por colisión con iones entrantes.
Factores de mejora de la señal en sustratos de SALDI
Dado que es la superficie activa responsable de la adsorción, desorción e ionización de las moléculas de analito la que caracteriza la técnica, la química superficial del material sustrato es sin duda crucial para el desempeño de SALDI. Pero es bastante difícil sacar una conclusión general debido a que la afinidad entre diferentes clases de sustratos y analitos es considerablemente versátil. Básicamente, la interacción entre esos dos componentes tiene un impacto en la captura y liberación de las moléculas de analito, así como el estado electrónico de la superficie del sustrato y la coeficiencia de transferencia de energía.
Otro aspecto importante son las propiedades físicas de los sustratos que podrían alterar directamente el proceso de desorción e ionización, especialmente para la vía térmicamente activada. Esto está estrechamente relacionado con el rápido aumento de la temperatura en la superficie del sustrato. Esas propiedades incluyen la coeficiencia de absorción óptica, la capacidad térmica y la conductividad térmica (o tasa de difusión de calor). Primero, una mayor coeficiencia de absorción óptica permite que el sustrato absorba y genere más calor cuando la fuente láser proporciona cierta cantidad de energía. Además, una menor capacidad calorífica generalmente conduce a un aumento mayor de la temperatura con la misma cantidad de calor. Además, una conductividad de audición más baja ayuda al sustrato a mantener una temperatura alta que dará como resultado un pico de temperatura más alto. Por lo tanto, la desorción térmica y la ionización podrían ocurrir de manera más rápida y efectiva.
Instrumentación
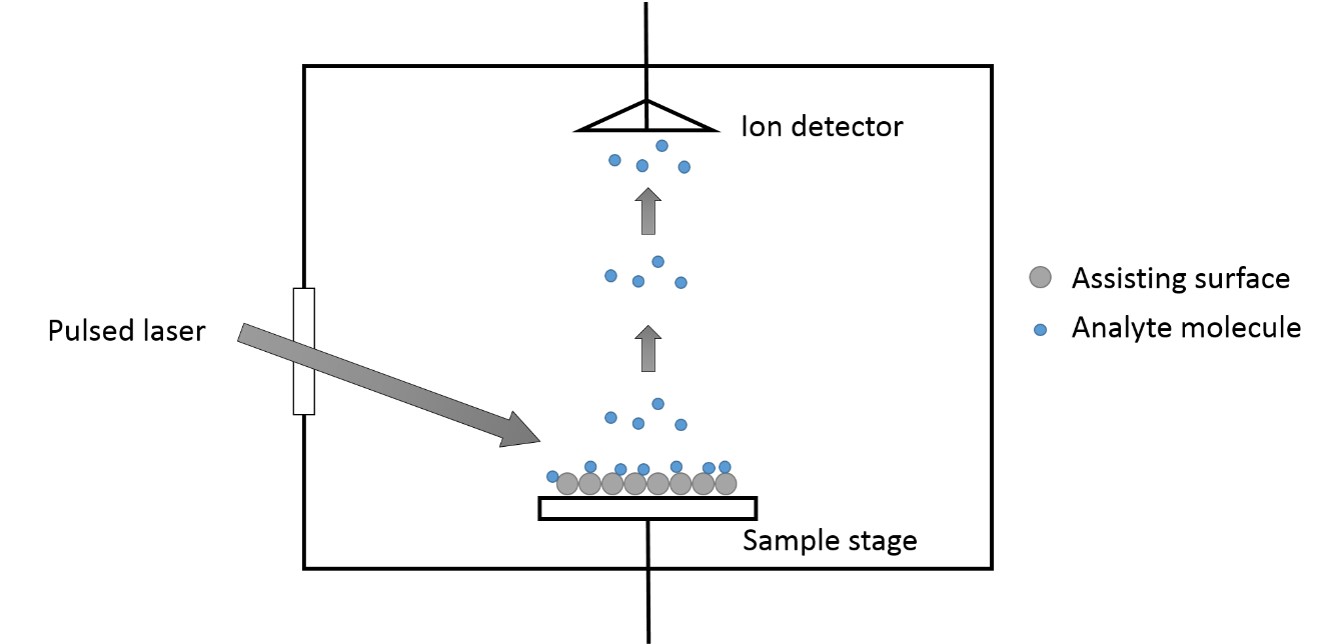
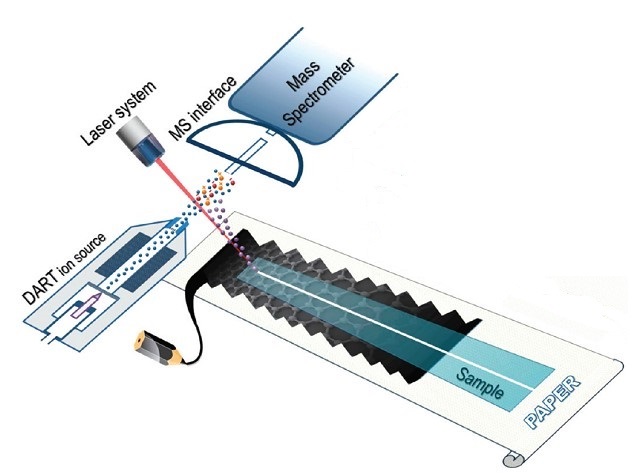
El instrumento involucrado en la SALDI que se muestra en la Figura\(\PageIndex{34}\) es similar al de MALDI en gran medida. Contiene una fuente láser que genera láser pulsado que excita la mezcla de muestra. Hay una etapa de muestra que coloca la mezcla de muestra de materiales de sustrato y analitos. Por lo general, el analizador de masas y el detector de iones están del otro lado para permitir que los iones pasen y se separen y detecten en base a diferentes valores de m/z. Se han logrado avances recientes que incorporan el análisis directo en tiempo real (DART) fuente de iones en el sistema SALDI-MS lo que permite realizar el análisis en condiciones ambientales. La figura\(\PageIndex{35}\) muestra su método SALDI-MS ambiental.
Ejemplos de nanomateriales utilizados para el análisis de analitos LMW en SALDI-MS
Silicio poroso como material de sustrato
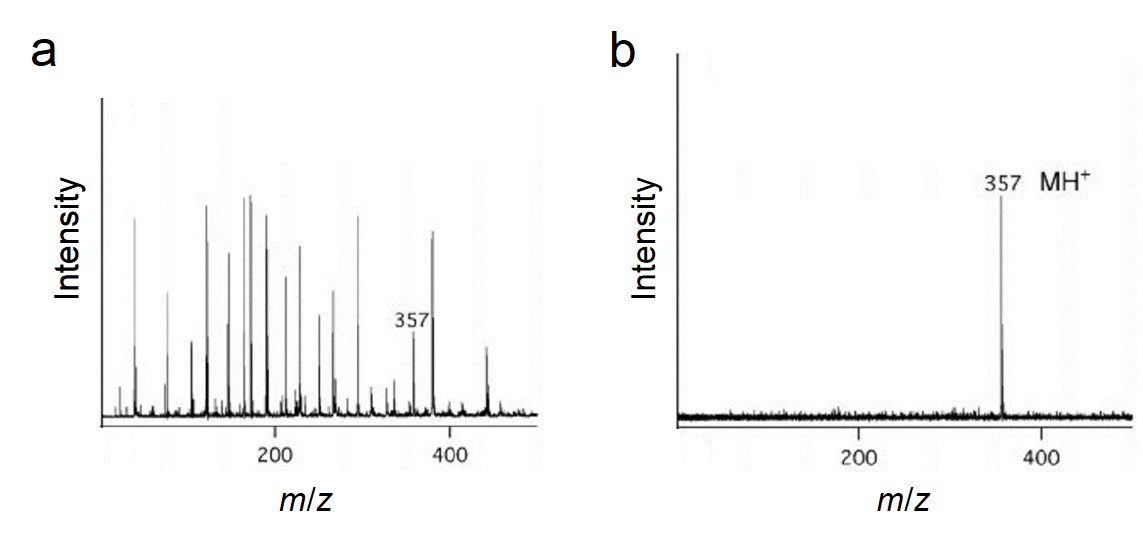
El silicio poroso con gran área de superficie podría usarse para atrapar ciertas moléculas de analito para el proceso de desorción e ionización sin matriz. Más interesante, se encontró una gran coeficiencia de absorción ultravioleta para este material poroso, lo que también mejoró el rendimiento de ionización. Se ha reportado que el uso de silicio poroso como sustrato en SALDI-MS fue capaz de trabajar a niveles de femtomol y attomol de analitos incluyendo como péptidos, cafeína, una molécula de fármaco antiviral (WIN), reserpina y N-octil - β -D-glucopiranósido. En comparación con MALDI-MS convencional, el DIOS-MS (que fue el tipo específico de SALDI en esta investigación) eliminó con éxito la interferencia de la matriz y mostró un pico cuasimolecular mucho mayor (MH +), lo que pudo observarse en la Figura\(\PageIndex{36}\). Además, la modificación química del silicio poroso pudo optimizar aún más las características de ionización.
Grafeno como material de superficie
El grafeno es un tipo de nanomaterial de carbono popular descubierto en 2004. Tiene una gran superficie que podría unir eficazmente las moléculas de analito. Por otro lado, la eficiencia de la desorción/ionización para analitos sobre una capa de grafeno se puede mejorar por su estructura monocapa simple y propiedades electrónicas únicas. Los compuestos polares que incluyen aminoácidos, poliaminas, fármacos anticancerosos y nucleósidos se pueden analizar con éxito. Además, las moléculas no polares pueden analizarse con alta resolución y sensibilidad debido a la naturaleza hidrofóbica del propio grafeno. Comparado con una matriz convencional, el grafeno presentó una alta eficiencia de desorción/ionización para compuestos no polares. El sustrato de grafeno funciona como un sustrato para atrapar analitos, y transfiere energía a los analitos tras la irradiación láser, lo que permite que los analitos sean fácilmente desorbidos/ionizados y la interferencia de la matriz sea eliminada. Se ha demostrado que el uso del grafeno como material sustrato evita la fragmentación de analitos y proporciona una buena reproducibilidad y una alta tolerancia a la sal, subrayando la posible aplicación del grafeno como matriz para el análisis MALDI-MS de muestras prácticas en matrices de muestra complejas. También se demuestra que el uso del grafeno como adsorbente para la extracción en fase sólida del escualeno podría mejorar en gran medida el límite de detección.
Combinación con GC
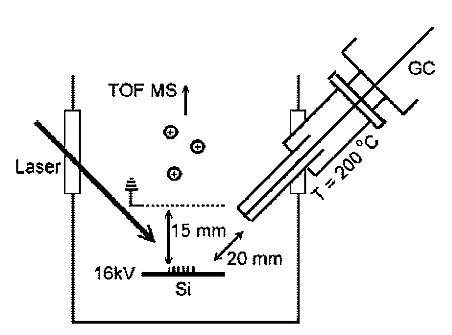
El análisis de SALDI-MS en fase gaseosa tiene una eficiencia de ionización relativamente alta, lo que conduce a una alta sensibilidad. En 2009, la cromatografía de gases (GC) se utilizó por primera vez con SALDI-MS, donde el sustrato de SALDI fue silicio amorfo y el analito fue feniletilaminas N-alquiladas. Los límites de detección estuvieron en el rango de los attomoles, pero se esperan mejoras en el futuro. Se espera que la combinación con GC amplíe aún más el uso de SALDI-MS que SALDI podría aplicarse a la separación e identificación de muestras con mayor complejidad. La configuración instrumental se muestra en la Figura\(\PageIndex{37}\).
Espectrometría de Masas Electroquímica Diferencial
En el estudio de la electroquímica, siempre había sido un reto obtener la detección inmediata y continua de productos electroquímicos debido a la limitada formación en la superficie del electrodo, hasta el descubrimiento de la espectrometría de masas electroquímica diferencial. Los científicos inicialmente probaron la idea combinando membrana porosa y espectrometría de masas para el análisis de productos en el estudio de la generación de oxígeno a partir de HClO 4 usando electrodo poroso en 1971. En 1984, se realizó otro experimento similar utilizando una membrana porosa de Teflon con 100 μm de lacas en la superficie entre los electrolitos y el sistema de vacío. Comparando con el experimento anterior, este experimento ha demostrado un sistema de vacío con derivado de tiempo mejorado que mostró detección casi inmediata de productos de reacción electroquímica volátiles, con alta sensibilidad de detección tan pequeña como “una monocapa” en el electrodo. En resumen, el experimento demostrado en 1984 no solo mostró detección continua de muestras en espectrometría de masas sino también las tasas de formación, que se distinguió de la técnica realizada anteriormente en 1971. De ahí que este método se denominara espectrometría de masas electroquímica diferencial (DEMS). Durante las últimas dos décadas, esta técnica ha evolucionado del uso del electrodo clásico al electrodo de disco giratorio (RDE), lo que proporciona un transporte más homogéneo y rápido de las especies de reacción a la superficie del electrodo.
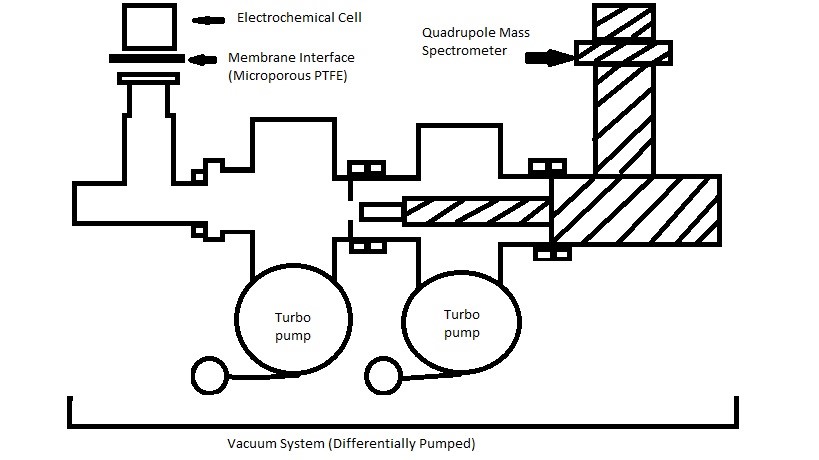
Descrita en términos básicos, la espectrometría de masas electroquímica diferencial es una técnica de caracterización que analiza especímenes utilizando tanto la experimentación electroquímica de media celda como la espectrometría de masas. Utiliza membrana no humectante para separar el electrolito acuoso y el electrolito gaseoso, cuyo electrolito gaseoso penetrará a través de la membrana y será ionizado y detectado en el espectrómetro de masas utilizando un sistema de vacío continuo de dos etapas. Este método analítico puede detectar reactivos electroquímicos gaseosos o volátiles, productos de reacción e incluso intermedios de reacción. El instrumento consta de tres componentes principales: semi-celda electroquímica, interfaz de membrana de PTFE (politetrafluoroetileno) y espectrómetro de masas cuadrupolo (QMS), que forma parte del sistema de vacío.
Operaciones DEMS
Todo el conjunto del instrumento se muestra en la Figura\(\PageIndex{38}\), que consta de tres componentes principales: una semicelda electroquímica, una interfaz de membrana de PTFE y el espectrómetro de masas cuadrupolo. En esta sección se explicará cada componente y se explorará su funcionalidad, y se proporcionará información adicional al final de esta sección. La membrana de PTFE es una membrana microporosa que separa el electrolito acuoso del electrolito volátil que será arrastrado a la porción de alto vacío. Usando la succión de alto vacío, se permitirá que las especies gaseosas o volátiles permeen a través de la membrana usando presión diferencial, dejando los materiales acuosos en la superficie debido a la naturaleza hidrofóbica de la membrana. La selección del material de membrana es muy importante para mantener tanto la hidrofobicidad como la difusión adecuada de especies volátiles. Las especies permeadas al SGC serán monitoreadas y medidas, y la cinética de formación se determinará al final. Dependiendo de las condiciones de funcionamiento, es posible que se requieran diferentes bombas de vacío.
Celdas Electroquímicas
El primer componente importante del instrumento DEMS es el diseño de celdas electroquímicas. Hay muchos diseños diferentes que se han desarrollado durante las últimas décadas, dependiendo de los tipos de reacciones electroquímicas, los tipos y tamaños de los electrodos. No obstante, en este capítulo solo se discutirá la celda clásica.
El método DEMS se demostró por primera vez usando el método clásico. En la Figura se muestra una configuración convencional de celda electroquímica\(\PageIndex{39}\). El material del electrodo en polvo se deposita sobre la membrana porosa para formar el electrodo de trabajo, mostrado como Material del electrodo de trabajo en la Figura\(\PageIndex{39}\). En la demostración de Wolber y Heitbaum, el electrodo se preparó teniendo pequeñas partículas de Pt depositadas sobre la membrana pintando una laca. Posteriormente en otras experimentaciones evolucionó para utilizar capa de electrocatalizador de pulverización catalizadora para una superficie más homogénea. El electrolito de celda acuosa está blindado con un cuerpo de vidrio boca abajo con apertura de túnel vertical a la membrana de PTFE. El material del electrodo de trabajo se encuentra por encima de la membrana de PTFE, donde es soportado mecánicamente por frita de acero inoxidable dentro de la brida de vacío. Tanto el material del electrodo de trabajo como la membrana de PTFE se comprimen entre las fundiciones al vacío y el espaciador de PTFE, que es un anillo que evita que el electrolito se escape. El contraelectrodo (CE) y el electrodo de referencia (RE) hechos de alambre de platino se colocan en la parte superior del material del electrodo de trabajo para crear el contacto eléctrico. Una de las principales ventajas del diseño clásico es el rápido tiempo de respuesta, con alta eficiencia de “0.5 para laca y 0.9 con el electrodo de pulverización catódica”. Sin embargo, este método plantea ciertas dificultades. Primero, los materiales electrolíticos serán absorbidos en el electrodo de trabajo antes de que penetre a través de la membrana. Debido a la limitación de la tasa de absorción, la concentración en la superficie del electrodo será menor que el volumen. Segundo, el electrolito volátil acuoso debe ser absorbido sobre el electrodo de trabajo, y luego seguido de evaporación a través de la membrana. Por lo tanto, la diferencia en las tasas de absorción y evaporación creará un cambio en el equilibrio. En tercer lugar, este método también se limita a los tipos de material que se pueden depositar en la superficie, como cristales simples o incluso algunas superficies de electrodos policristalinos. Por último, la forma en que el RE es la posición podría potencialmente introducir impurezas en el sistema, lo que interferirá con el experimento.
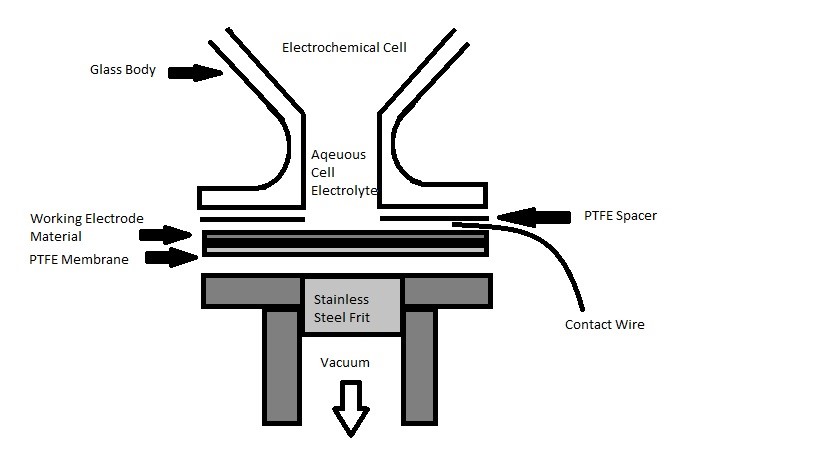
Interfaz de Membrana
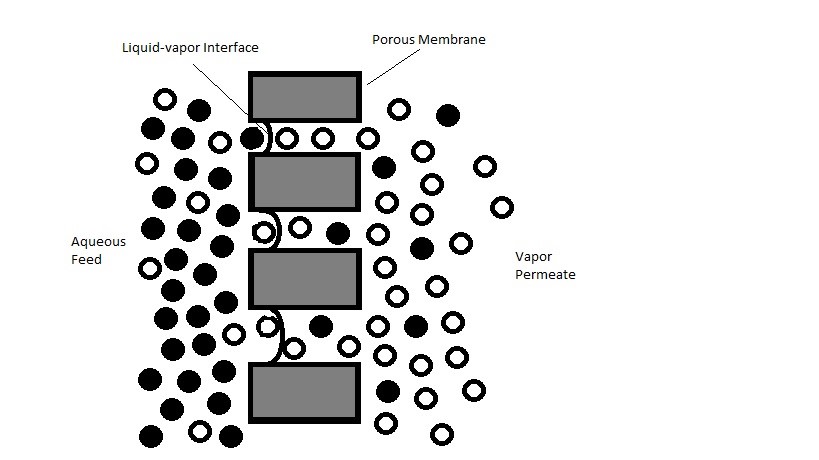
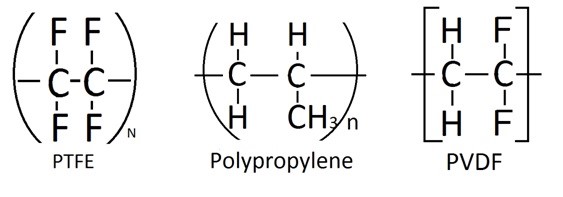
La membrana de PTFE se coloca entre la celda de electrolito acuoso y el sistema de alto vacío en el otro extremo. Actúa como una barrera que impide el paso del electrolito acuoso, mientras que su selectividad permite que las especies electroquímicas vaporizadas se transporten al lado de alto vacío, el cual el proceso es similar a la destilación por membrana al vacío que se muestra en la Figura\(\PageIndex{41}\). Para evitar que la solución acuosa penetre a través de la membrana, la superficie de la membrana debe ser hidrófoba, que es una propiedad material que repele el agua o el fluido acuoso. Por lo tanto, en cada ubicación de poro, hay una interfaz de vapor y líquido donde el líquido permanecerá en la superficie mientras que el vapor penetrará en la membrana. Entonces el transporte del material en fase vapor se desencadena por la diferencia de presión creada a partir del vacío en el otro extremo de la membrana. Por lo tanto, el tamaño del poro es crucial para controlar sus propiedades hidrófobas y la velocidad de transferencia a través de la membrana. Cuando el tamaño de poro es inferior a 0.8 μm, se activa la propiedad hidrofóbica. Este número se determina calculando la tensión superficial del líquido, el ángulo de contacto y la presión aplicada. Por lo tanto, se desea una membrana con tamaños de poro relativamente pequeños y distribución de poros grandes. En general, los materiales de membrana utilizados son “típicamente 0.02 μm de tamaño con espesor entre 50 y 110 μm”. En cuanto a materiales, se han probado otros materiales como polipropileno y fluoruro de polivinilideno (PVDF) (Figura\(\PageIndex{41}\)); sin embargo, el material PTFE (Figura\(\PageIndex{42}\)) como membrana ha demostrado una mejor durabilidad y resistencia química al ambiente electroquímico. Por lo tanto, se muestra que el PTFE es el mejor candidato para dicha aplicación, y generalmente se lamina sobre polipropileno para mejorar las propiedades mecánicas. A pesar de la propiedad hidrofóbica del material PTFE, una cantidad significativa de material acuoso penetra a través de la membrana debido a la gran caída de presión. Por lo tanto, el correcto dimensionamiento de las bombas de vacío es crucial para mantener el flujo de gas a transportar a la espectrometría de masas a la presión deseada. Se discutirá más información sobre el sistema de vacío. Además, se ha utilizado capilar en reemplazo de la membrana; sin embargo, este método no se discutirá aquí.
Vacío y QMS
El sistema de vacío de tamaño correcto puede garantizar la máxima cantidad de material de vapor a transportar a través de la membrana. Cuando la caída de presión no es adecuada, parte del material de vapor puede quedar en el lado acuoso como se muestra en la Figura\(\PageIndex{43}\). Sin embargo, cuando la caída de presión es demasiado grande, se extraerá demasiado electrolito acuoso de la interfaz líquido-vapor, aumentando posteriormente la carga en las bombas de vacío. En los casos de bombas de tamaño incorrecto puede reducir la eficiencia de la bomba y reducir la vida útil de la bomba si dicho problema no se corrige de inmediato. Además, para que la espectrometría de masas funcione correctamente, el flujo de gas deberá mantenerse en cierto flujo. Por lo tanto, las bombas de vacío deben proporcionar un flujo constante de gas alrededor de 0.09 mbar/s.cm2 consistente principalmente con especies gaseosas o volátiles y otras especies que serán enviadas a espectrometría de masas para su análisis. Además, debido a la limitación de la velocidad de la bomba de una sola bomba de vacío, se necesitará un sistema de vacío con dos o más bombas. Por ejemplo, si se requiere 0.09 mbar/s.cm 2 y una velocidad de bombeo de 300 s -1 que opera a 10 -5 mbar, el área geométrica aceptable de la membrana es 0.033 cm -2. Para aumentar el área de la membrana, se requerirán bombas de adición para lograr el mismo flujo de gas.
Información Adicional
Existen varias otras técnicas analíticas como la voltametría cíclica, el paso potencial y el paso galvánico que se pueden combinar con el experimento DEMS. La voltametría cíclica puede proporcionar resultados cuantitativos y cualitativos utilizando la dependencia potencial. Como resultado, tanto la corriente iónica de las especies interesadas como la corriente de electrodo faradaica (la corriente generada por la reducción u oxidación de alguna sustancia química en un electrodo) se registrarán al combinar voltamperometría cíclica y DEMS.
Aplicaciones
La falta de comercialización de esta técnica la ha limitado únicamente a la investigación académica. El mayor campo de aplicación de DEMS está en las reacciones electrocatalíticas. Además, también se utiliza investigación de celdas de combustible, reacciones de desintoxicación, sensores de gas electroquímicos o investigaciones más relevantes fundamentales como descomposición de líquidos iónicos etc.
Espectrometría de masas electroquímica diferencial de pila de combustible: Electrooxidación de etanol
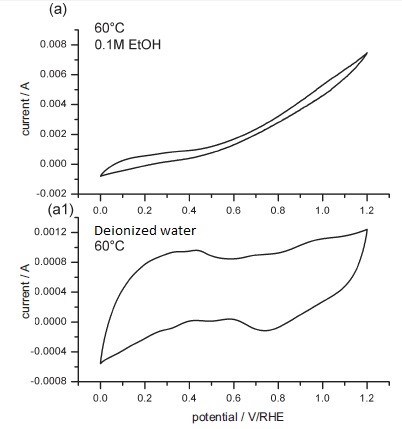
La reacción de oxidación de etanol se estudió mediante ensambles de electrodos de membrana alcalina (MEA), construidos con catalizador de nanopartículas de Pt y membrana polimérica alcalina. DEMS se utilizará para estudiar la mecánica de la reacción de oxidación de etanol en los catalizadores basados en pt. Los productos relevantes de la reacción de oxidación son dióxido de carbono, acetaldehído y ácido acético. Sin embargo, tanto el dióxido de carbono como el acetaldehído tienen el mismo peso molecular, el cual es de 44 g/mol. Un enfoque es monitorear los fragmentos principales donde se utilizaron CO 2 2+ ionizado a m/z = 22 y COH + a m/z = 29. La espectrometría de masas electroquímica diferencial puede detectar productos volátiles de la reacción electroquímica; sin embargo, las detecciones pueden variar por solubilidad o punto de ebullición. El CO 2 es muy volátil, pero también soluble en agua. Si KOH está presente, DEMS no detectará ningún rastro de CO 2. Por lo tanto, todas las impurezas alcalinas adicionales deben eliminarse antes de tomar las medidas. Las características electroquímicas también se pueden medir bajo diversas condiciones y ejemplos mostrados en la Figura\(\PageIndex{43}\). Además, el CCE (eficiencia de corriente de CO 2) se midió bajo diferentes potenciales. Usando el CCE, el estudio concluyó que el etanol sufre una oxidación más completa usando MEA alcalino que MEA ácido.
Estudios sobre la Descomposición de Líquidos Iónicos
Los líquidos iónicos (IL) tienen varias propiedades como alta conductividad iónica, baja presión de vapor, alta estabilidad térmica y electroquímica, lo que los convierte en un gran candidato para el electrolito de la batería. Por lo tanto, es importante tener una mejor comprensión de la estabilidad de la reacción y de los productos formados durante el comportamiento de descomposición. El DEMS es un método poderoso donde puede proporcionar detección en línea de los productos volátiles; sin embargo, se encuentra con problemas con alta viscosidad de ILs y baja permeabilidad debido al tamaño de las moléculas. Por lo tanto, los investigadores modificaron la configuración tradicional de los DEMS, que el método modificado hizo uso de la baja presión de vapor de los ILs y tienen celda electroquímica colocada directamente en el sistema de vacío. Este experimento muestra que esta técnica puede diseñarse para una aplicación muy específica y se puede modificar fácilmente.
Conclusión
La técnica DEMS puede proporcionar detección en línea de productos para reacciones electroquímicas tanto analítica como cinéticamente. Además, los resultados se entregan con alta sensibilidad donde tanto los productos como los subproductos pueden ser detectados siempre y cuando sean volátiles. Se puede ensamblar fácilmente en el ambiente de laboratorio. Durante las últimas décadas, esta técnica ha demostrado un desarrollo avanzado y ha brindado buenos resultados para muchas aplicaciones como celdas de combustible, sensores de gas etc. Sin embargo, esta técnica tiene su limitación. Hay muchos factores que deben tenerse en cuenta al diseñar este sistema como la reacción electroquímica de media celda, la velocidad de absorción y etc. Debido a estas limitaciones, se debe seleccionar el tipo de membrana y se debe dimensionar la bomba en consecuencia. Por lo tanto, este método de caracterización no es de talla única y necesitará ser modificado en base a los parámetros experimentales. Por lo tanto, el siguiente paso de desarrollo para el DEMS no es sólo mejorar sus funciones, sino también ser utilizado más allá del laboratorio académico.