
7.2: Estructuras de Semiconductores de Elementos y Compuestos
- Page ID
- 70991

Un solo cristal de un semiconductor elemental (por ejemplo, silicio) o compuesto (por ejemplo, arseniuro de galio) forma la base de casi todos los dispositivos semiconductores. La capacidad de controlar las propiedades electrónicas y optoelectrónicas de estos materiales se basa en una comprensión de su estructura. Además, los metales y muchos de los aislantes empleados dentro de un dispositivo microelectrónico también son cristalinos.
Grupo IV (14) Elementos
Cada una de las fases semiconductoras de los elementos del grupo IV (14), C (diamante), Si, Ge y α-SN, adoptan la estructura cúbica del diamante (Figura\(\PageIndex{1}\)). Sus constantes de celosía (a, Å) y densidades (ρ, g/cm3) se dan en la Tabla\(\PageIndex{1}\).

| Element | Parámetro de celosía, a (Å) | Densidad (g/cm 3) |
|---|---|---|
| carbono (diamante) | 3.56683 (1) | 3.51525 |
| silicio | 5.4310201 (3) | 2.319002 |
| germanio | 5.657906 (1) | 5.3234 |
| estaño (α-SN) | 6.4892 (1) |
Como se esperaría el aumento del parámetro de celosía en el orden C < Si < Ge < α-SN. El silicio y el germanio forman una serie continua de soluciones sólidas con parámetros que varían gradualmente. Cabe destacar el alto grado de precisión que los parámetros de celosía son conocidos para los cristales de alta pureza de estos elementos. Además, es importante señalar la temperatura a la que se realizan las mediciones estructurales, ya que los parámetros de celosía son dependientes de la temperatura (Figura\(\PageIndex{1}\)). La constante reticular (a), en Å, para silicio de alta pureza puede calcularse para cualquier temperatura (T) en el rango de temperatura 293 - 1073 K por la fórmula que se muestra a continuación.
\[ a_{T}\ =\ 5.4304\ +\ 1.8138 \times 10^{-5}\ (T- 298.15\ K)\ +\ 1.542 \times 10^{-9}\ (T-298.15\ K) \label{1} \]

Aunque las formas cúbicas de diamante de Si y Ge son las únicas formas de interés directo para los dispositivos semiconductores, cada una existe en numerosas formas cristalinas de alta presión y metaestables. Estos se describen junto con sus interconversiones, en la Tabla\(\PageIndex{2}\).
| Fase | Estructura | Observaciones |
|---|---|---|
| Si I | diamante cúbico | estable a presión normal |
| Si II | estructura de hojalata gris | formado a partir de Si I o Si V por encima de 14 GPa |
| Si III | cúbico | metaestable, formado a partir de Si II por encima de 10 GPa |
| Si IV | hexagonal | |
| Si V | no identificado | estable por encima de 34 GPa, formado a partir de Si II por encima de 16 GPa |
| Si VI | hexagonal, cierre, embalado | estable por encima de 45 GPa |
| Ge I | diamante cúbico | fase de baja presión |
| Ge II | Estructura β-estaño | formado a partir de Ge I por encima de 10 GPa |
| Ge III | tetragonal | formado por temple Ge II a baja presión |
| Ge IV | centrado en el cuerpo | formado por temple Ge II a 1 atm a 200 K |
Compuestos del Grupo III-V (13-15)
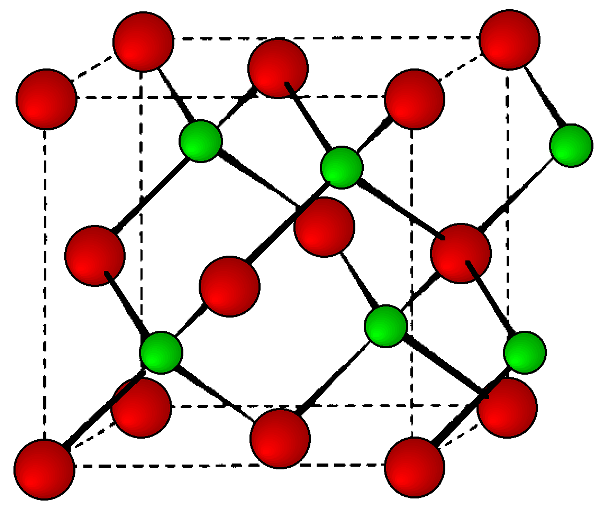
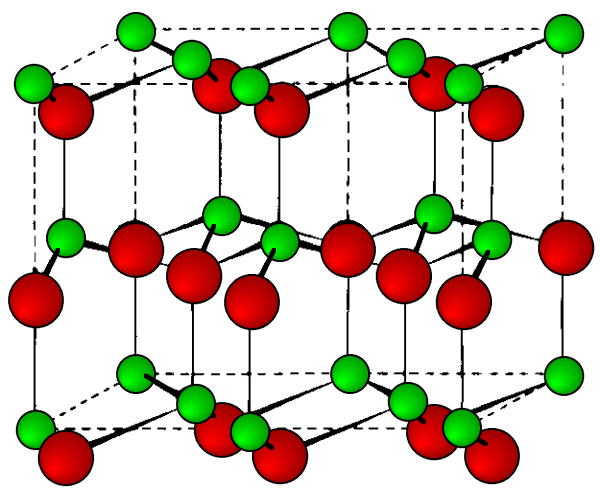
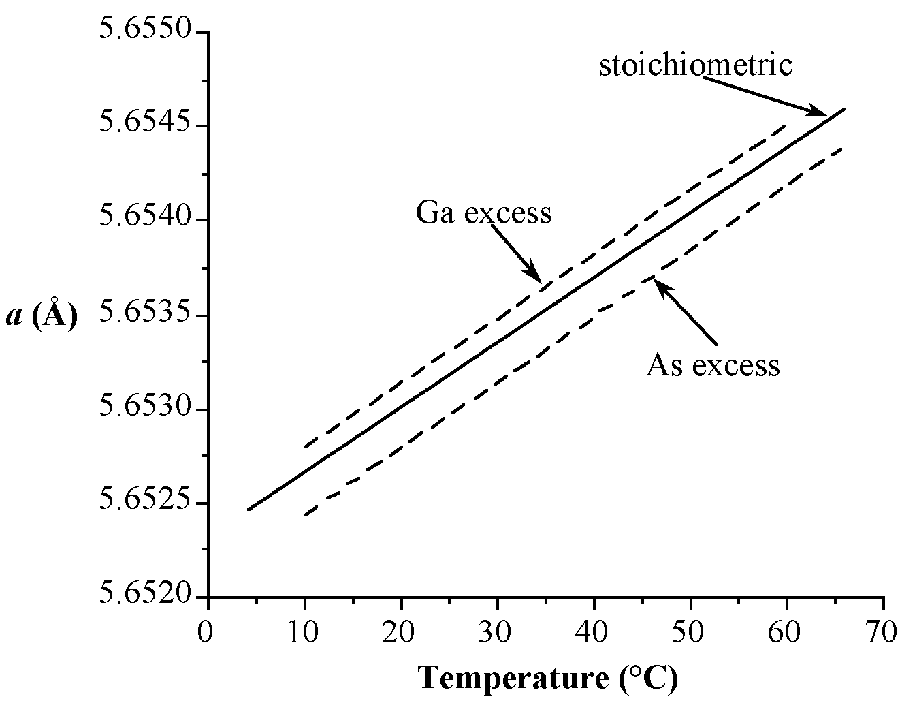
Las fases estables para los arsenidos, fosfuros y antimonuros de aluminio, galio e indio presentan estructuras de blenda de zinc (Figura\(\PageIndex{3}\)). En contraste, los nitruros se encuentran como estructuras de wurtzita (por ejemplo, Figura\(\PageIndex{4}\)). La estructura, los parámetros reticulares y las densidades de los compuestos III-V se dan en la Tabla\(\PageIndex{3}\). Cabe señalar que contrariamente a lo esperado el parámetro reticular de los compuestos de galio es menor que su homólogo de aluminio; para GaAs a = 5.653 Å; AlAs a = 5.660 Å. Al igual que con los elementos del grupo IV, los parámetros de la red son altamente dependientes de la temperatura; sin embargo, la variación adicional surge de cualquier desviación de la estequiometría absoluta. Estos efectos se muestran en la Figura\(\PageIndex{4}\).
| Compuesto | Estructura | Parámetro de celosía (Å) | Densidad (g/cm 3) |
|---|---|---|---|
| AIN | wurtzita | a = 3.11 (1), c = 4.98 (1) | 3.255 |
| AIP | blenda de zinc | a = 5.4635 (4) | 2.40 (1) |
| AIA | blenda de zinc | a= 5.660 | 3.760 |
| AISb | blenda de zinc | a = 6.1355 (1) | 4.26 |
| GaN | wurtzita | a = 3.190, c=5.187 | |
| GaP | blenda de zinc | a= 5.4505 (2) | 4.138 |
| GaAs | blenda de zinc | a= 5.56325 (2) | 5.3176 (3) |
| INn | wurtzita | a= 3.5446, c= 5.7034 | 6.81 |
| InP | blenda de zinc | a= 5.868 (1) | 4.81 |
| INAs | blenda de zinc | a= 6.0583 | 5.667 |
| InsB | blenda de zinc | a= 6.47937 | 5.7747 (4) |
La homogeneidad de estructuras de aleaciones para una amplia gama de soluciones sólidas que se formarán entre compuestos III-V en casi cualquier combinación. Se forman dos clases de aleaciones ternarias: III x -III 1-x -V (por ejemplo, Al x -Ga 1-x -As) y III-V 1-x -V x (por ejemplo, Ga-As 1-x -P x). Mientras que las aleaciones cuaternarias del tipo III x -III 1-x -V y -V 1-y permiten el crecimiento de materiales con parámetros similares de celosía, pero un amplio rango de huecos de banda. Una aleación ternaria muy importante, especialmente en aplicaciones optoelectrónicas, es Al x -Ga 1-x -As y su parámetro reticular (a) está directamente relacionado con la composición (x).
\[ a\ =\ 5.6533\ +\ 0.0078\ x \nonumber \]
No todos los compuestos III-V tienen fases de alta presión bien caracterizadas. Sin embargo, en cada caso donde se observa una fase de alta presión el número de coordinación tanto del elemento del grupo III como del grupo V aumenta de cuatro a seis. Así, AlP experimenta una transformación de mezcla de zinc a sal de roca a alta presión por encima de 170 kbar, mientras que AlSb y GaAs forman estructuras ortorrómbicas distorsionadas de sal rocosa por encima de 77 y 172 kbar, respectivamente. Se propone una estructura ortorrómbica para la forma de alta presión de InP (>133 kbar). El arseniuro de indio (INAs) sufre transformaciones de dos fases. La estructura de la mezcla de zinc se convierte en una estructura de sal de roca por encima de 77 kbar, que a su vez forma una estructura β-estaño por encima de 170 kbar.
Compuestos del Grupo II-VI (12-16)
Las estructuras de los semiconductores compuestos II-VI son menos predecibles que las de los compuestos III-V (anteriores), y aunque la estructura de blenda de zinc existe para casi todos los compuestos, existe una tendencia más fuerte hacia la forma hexagonal de wurtzita. En varios casos la estructura de la mezcla de zinc se observa bajo condiciones ambientales, pero puede convertirse a la forma de wurtzita al calentarse. En general la forma wurtzita predomina con los aniones más pequeños (e.g., óxidos), mientras que la blenda de zinc se convierte en la fase más estable para los aniones más grandes (por ejemplo, telururos). Una excepción es el sulfuro de mercurio (HG) que es el arquetipo para la fase trigonal de cinabrio.En la tabla se\(\PageIndex{5}\) enumera la fase estable de los calcogenidos de zinc, cadmio y mercurio, junto con sus fases de alta temperatura cuando corresponda. Las soluciones sólidas de los compuestos II-VI no se forman tan fácilmente como para los compuestos III-V; sin embargo, dos ejemplos importantes son ZnS x Se 1-x y Cd x Hg 1-x Te.
| Compuesto | Estructura | Parámetro de celosía (Å) | Densidad (g/cm 3) |
| ZnS | blenda de zinc | a= 5.410 | 4.075 |
| wurtzita | a = 3.822, c= 6.260 | 4.087 | |
| ZnSe | blenda de zinc | a = 5.668 | 5.27 |
| ZnTe | blenda de zinc | a = 6.10 | 5.636 |
| CDs | wurtzita | a = 4.136, c = 6.714 | 4.82 |
| CdSe | wurtzita | a = 4.300, c = 7.011 | 5.81 |
| CdTe | blenda de zinc | a = 6.482 | 5.87 |
| HgS | cinabrio | a = 4.149, c = 9.495 | |
| blenda de zinc | a = 5.851 | 7.73 | |
| HGse | blenda de zinc | a = 6.085 | 8.25 |
| HGTe | blenda de zinc | a = 6.46 | 8.07 |
Los calcogenuros de zinc se transforman en una estructura de cloruro de cesio bajo altas presiones, mientras que los compuestos de cadmio forman fases de alta presión de sal de roca (Figura\(\PageIndex{6}\)). El seleniuro de mercurio (HGSe) y el telururo de mercurio (HGTe) se convierten en la estructura arquetipo de sulfuro de mercurio, cinabrio, a alta presión.
I-III-VI 2 (11-13-16) Compuestos
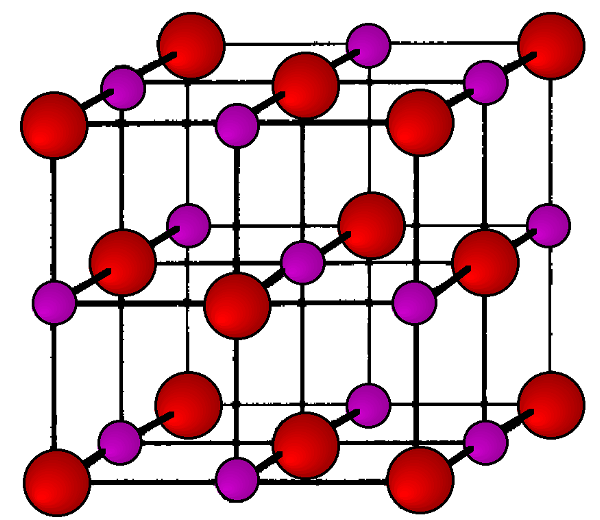
Casi todos los compuestos I-III-VI2 a temperatura ambiente adoptan la estructura de calcopirita (Figura\(\PageIndex{7}\)). Las constantes y densidades de celda se dan en la Tabla\(\PageIndex{6}\). Aunque hay pocos reportes de fases de alta temperatura o alta presión, AGINS2 ha demostrado existir como un polimorfo ortorrómbico de alta temperatura (a = 6.954, b = 8.264 y c = 6.683 Å), y AgInTe2 forma una fase cúbica a altas presiones.
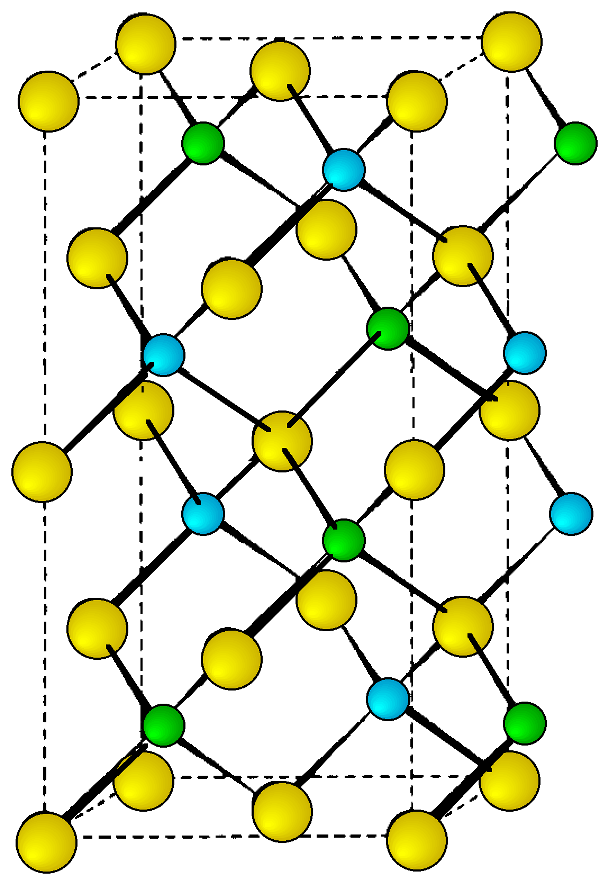
| Compuesto | Parámetro de celosía a (Å) | Parámetro de celosía c (Å) | Densidad (g cm 3) |
| CUALS 2 | 5.32 | 10.430 | 3.45 |
| CualSe 2 | 5.61 | 10.92 | 4.69 |
| CuAlte | 5.96 | 11.77 | 5.47 |
| CuGas 2 | 5.35 | 10.46 | 4.38 |
| CuGasa 2 | 5.61 | 11.00 | 5.57 |
| CuGate | 6.00 | 11.93 | 5.95 |
| CUINs 2 | 5.52 | 11.08 | 4.74 |
| CuinSE 2 | 5.78 | 11.55 | 5.77 |
| CuInte 2 | 6.17 | 12.34 | 6.10 |
| AGAL 2 | 6.30 | 11.84 | 6.15 |
| AGGas 2 | 5.75 | 10.29 | 4.70 |
| AGGasa 2 | 5.98 | 10.88 | 5.70 |
| AGgate | 6.29 | 11.95 | 6.08 |
| AGINs | 5.82 | 11.17 | 4.97 |
| AGinSE 2 | 6.095 | 11.69 | 5.82 |
| Aginte 2 | 6.43 | 12.59 | 6.96 |
De los compuestos I-III-VI2, los calcogenidos de cobre indio (CuINe2) son sin duda los más estudiados para su aplicación en células solares. Una de las ventajas de los compuestos de calcogenuro de indio y cobre es la formación de soluciones sólidas (aleaciones) de fórmula Cuine2-xe'x, donde la variable de composición (x) varía de 0 a 2. También se han examinado los sistemas Cuins2-xSex y Cuinse2-xTex, así como el sistema cuaternario Cugayin1-YS2-xSex. Como se esperaría a partir de una consideración de los radios iónicos relativos de los calcogenidos, los parámetros reticulares de la aleación Cuins2-xSEX deberían aumentar con un mayor contenido de selenio. La ley de Vergard requiere que la constante reticular para una solución lineal de dos semiconductores varíe linealmente con la composición (e.g., como se observa para AlxGa1-xAs), sin embargo, la variación de las constantes de retícula tetragonal (a y c) con composición para Cuins2-xSX se describen mejor por las relaciones parabólicas.
\[ a\ =\ 5.532\ +\ 0.0801x\ +\ 0.026 x^{2} \nonumber \]
\[ c\ =\ 11.156\ +\ 0.1204x\ +\ 0.0611 x^{2} \nonumber \]
Se observa una relación similar para las aleaciones Cuinse2-xTex.
\[ a\ =\ 5.783\ +\ 0.1560 x\ +\ 0.0212x^{2} \nonumber \]
\[ c\ =\ 11.628\ +\ 0.3340x\ +\ 0.0277x^{2} \nonumber \]
La gran diferencia en radios iónicos entre S y Te (0.37 Å) impide la formación de soluciones sólidas en el sistema Cuins2-xTex, sin embargo, se ha reportado la aleación única Cuins1.5Te0.5.
Efectos de orientación
Una vez que se producen cristales individuales de silicio de alta pureza o arseniuro de galio, se cortan en obleas de tal manera que la cara expuesta de estas obleas sea los planos cristalográficos {100} o {111}. La estructura relativa de estas superficies es importante con respecto a la oxidación, el grabado y el crecimiento de película delgada. Estos procesos son sensibles a la orientación; es decir, dependen de la dirección en la que se corte la rodaja de cristal.
Densidad atómica y enlaces colgados
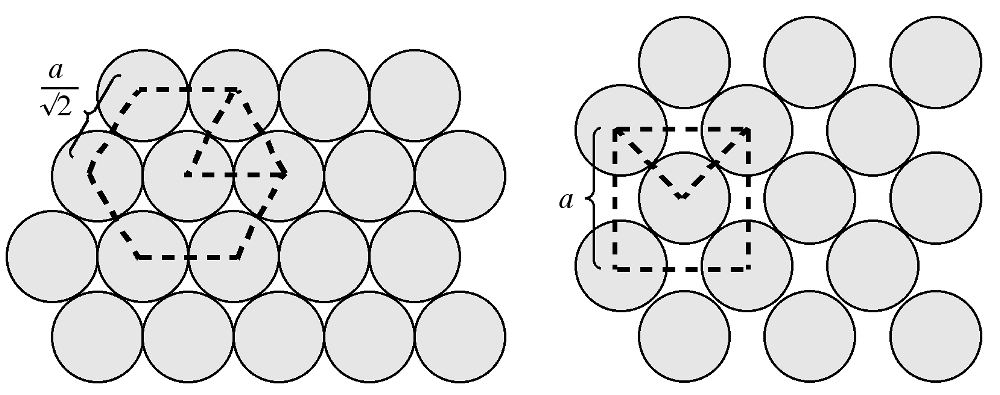
Los planos principales en un cristal pueden diferenciarse de varias maneras, sin embargo, la densidad de átomos y/o enlaces son útiles para predecir gran parte de la química de las superficies semiconductoras. Dado que tanto el arseniuro de silicio como el galio son estructuras fcc y las {100} y {111} son las únicas superficies tecnológicamente relevantes, las discusiones se limitarán a fcc {100} y {111}.
La densidad de átomos de una superficie puede definirse como el número de átomos por unidad de área. La figura muestra una vista esquemática de los planos {111} y {100} en una celosía fcc. El plano {111} consiste en una matriz hexagonal compacta en la que las direcciones del cristal dentro del plano están orientadas a 60° entre sí. El empaque hexagonal y la orientación de las direcciones del cristal se indican en la Figura\(\PageIndex{8}\) b como un hexágono superpuesto. Dado que la distancia interatómica intraplanar puede definirse como una función del parámetro de celosía, el área de este hexágono puede calcularse fácilmente. Por ejemplo en el caso del silicio, el hexágono tiene un área de 38.30 Å2. El número de átomos dentro del hexágono es tres: el átomo en el centro más 1/3 de cada uno de los seis átomos en los vértices del hexágono (cada uno de los átomos en los vértices de los hexágonos es compartido por otros tres hexágonos adyacentes). Así, se calcula que la densidad atómica del plano {111} es de 0.0783 Å-2. De manera similar, se puede calcular la densidad atómica del plano {100}. El plano {100} consiste en una matriz cuadrada en la que las direcciones del cristal dentro del plano están orientadas a 90° entre sí. Dado que el cuadrado coincide con una de las caras de la celda unitaria, el área del cuadrado puede calcularse fácilmente. Por ejemplo en el caso del silicio, el cuadrado tiene un área de 29.49 Å2. El número de átomos dentro del cuadrado es 2: el átomo en el centro más 1/4 de cada uno de los cuatro átomos en los vértices del cuadrado (cada uno de los átomos en las esquinas del cuadrado son compartidos por otros cuatro cuadrados adyacentes). Así, se calcula que la densidad atómica del plano {100} es de 0.0678 Å-2. Si bien estos valores para la densidad atómica son específicos para el silicio, su relación es constante para todas las estructuras cúbicas de diamante y de blenda de zinc: {100}: {111} = 1:1 .155. En general, cuanto menos enlaces colgados, más estable es una estructura superficial.
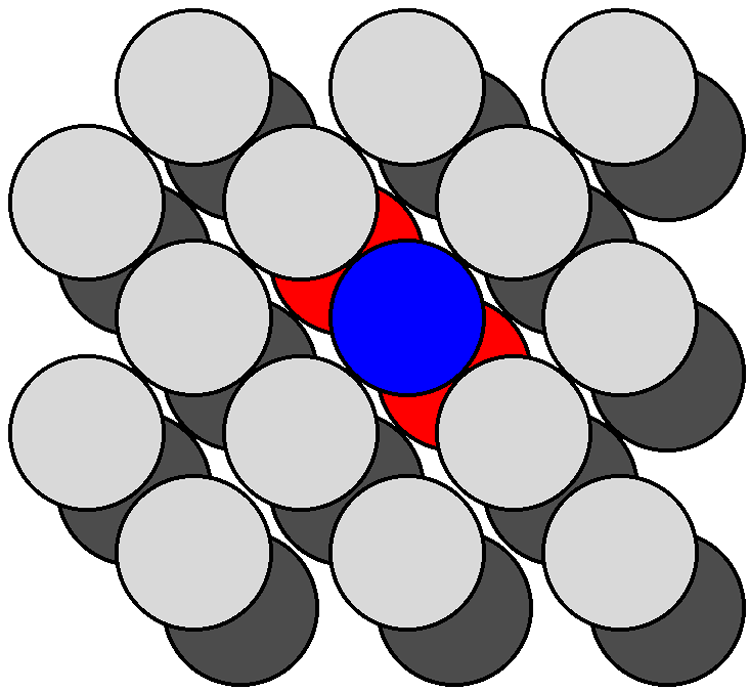
Un átomo dentro de un cristal de cualquier material tendrá un número de coordinación (n) determinado por la estructura del material. Por ejemplo, todos los átomos dentro de la masa de un cristal de silicio estarán en un ambiente tetraédrico de cuatro coordenadas (n = 4). Sin embargo, en la superficie de un cristal los átomos no harán su completo complemento de enlaces. Por lo tanto, cada átomo tendrá menos vecinos más cercanos que un átomo dentro del grueso del material. Los bonos faltantes se denominan comúnmente enlaces colgados. Aunque esta descripción no es particularmente precisa, es, sin embargo, ampliamente empleada y como tal se utilizará en la presente memoria. El número de enlaces colgados puede definirse como la diferencia entre el número de coordinación ideal (determinado por la estructura cristalina a granel) y el número de coordinación real observado en la superficie.
La figura\(\PageIndex{9}\) muestra una sección de las {111} superficies de una red cúbica de diamante vista perpendicular al plano {111}. Los átomos dentro de la masa tienen un número de coordinación de cuatro. En contraste, los átomos en la superficie (e.g., el átomo mostrado en azul en la Figura\(\PageIndex{10}\) están unidos cada uno a solo otros tres átomos (los átomos mostrados en rojo en la Figura), así cada átomo de superficie tiene un enlace colgante. Como se puede ver en la Figura\(\PageIndex{10}\), que muestra los átomos en la superficie {100} vista perpendicular al plano {100}, cada átomo en la superficie (por ejemplo, el átomo mostrado en azul en la Figura solo\(\PageIndex{9}\) se coordina con otros dos átomos (los átomos se muestran en rojo en la Figura\(\PageIndex{10}\), dejando dos enlaces colgando por átomo. Cabe señalar que se encuentra el mismo número de enlaces colgados para los planos {111} y {100} de una celosía de blenda de zinc. La proporción de enlaces colgados para los planos {100} y {111} de todas las estructuras cúbicas de diamante y blenda de zinc es {100}: {111} = 2:1. Además, dado que se conocen las densidades atómicas de cada plano entonces se determina que la relación de las densidades de enlace colgando es: {100}: {111} = 1:0 .577.

Silicio
Para el silicio, los planos {111} están más empaquetados que los planos {100}. Como resultado, el crecimiento de un cristal de silicio es por lo tanto más lento en la <111>dirección, ya que requiere colocar una capa atómica empaquetada cerca sobre otra capa en su forma empaquetada más cercana. Como consecuencia <111>Si es el más fácil de cultivar, y por lo tanto el menos costoso.
La disolución o grabado de un cristal está relacionada con el número de enlaces rotos ya presentes en la superficie: cuantos menos enlaces se rompan para eliminar un átomo individual de un cristal, más fácil será disolver el cristal. Como consecuencia de tener solo un enlace colgante (que requiere que se rompan tres enlaces) el grabado de silicio es más lento en la <111>dirección. Las propiedades electrónicas de una oblea de silicio también están relacionadas con el número de enlaces colgados.
Los microcircuitos de silicio generalmente se forman en una oblea de cristal único que se corta en cubitos después de la fabricación, ya sea aserrando parcialmente a través del grosor de la oblea o rayando (trazando) la superficie, y luego rompiendo físicamente. La rotura física de la oblea se produce a lo largo de los planos de escisión naturales, que en el caso del silicio son los planos {111}.
Arsenuro de galio
La red de blenda de zinc observada para el arseniuro de galio resulta en consideraciones adicionales sobre la del silicio. Aunque el plano {100} de GaAs es estructuralmente similar al del silicio, existen dos posibilidades: una cara que consiste en todos los átomos de galio o todos los átomos de arsénico. En cualquier caso los átomos superficiales tienen dos enlaces colgando, y las propiedades de la cara son independientes de si la cara es galio o arsénico.
El plano {111} también tiene la posibilidad de constar de todo el galio o todo el arsénico. Sin embargo, a diferencia de los {100} planos hay una diferencia significativa entre las dos posibilidades. La figura\(\PageIndex{11}\) muestra la estructura de arseniuro de galio representada por dos redes fcc interpenetrantes. El eje [111] es vertical dentro del plano de la página. Aunque la estructura consiste en capas alternas de galio y arsénico apiladas a lo largo del eje [111], la distancia entre las capas sucesivas alterna entre grandes y pequeñas. Al asignar arsénico como celosía padre, el orden de las capas en la dirección [111] es As-Ga-As-Ga-As-Ga, mientras que en la dirección [111] las capas están ordenadas, Ga-As-Ga-As-Ga-As (Figura\(\PageIndex{11}\)) .En silicio estas dos direcciones son, por supuesto, idénticas. La superficie de un cristal sería arsénico, con tres enlaces colgando, o galio, con un enlace colgando. Claramente, este último es energéticamente más favorable. Así, el plano (111) que se muestra en la Figura\(\PageIndex{11}\) se denomina cara (111) Ga. Por el contrario, el plano [111] sería galio, con tres enlaces colgados, o arsénico, con un enlace colgante. Nuevamente, este último es energéticamente más favorable y el plano [111] es por lo tanto llamado el (111) Como cara.
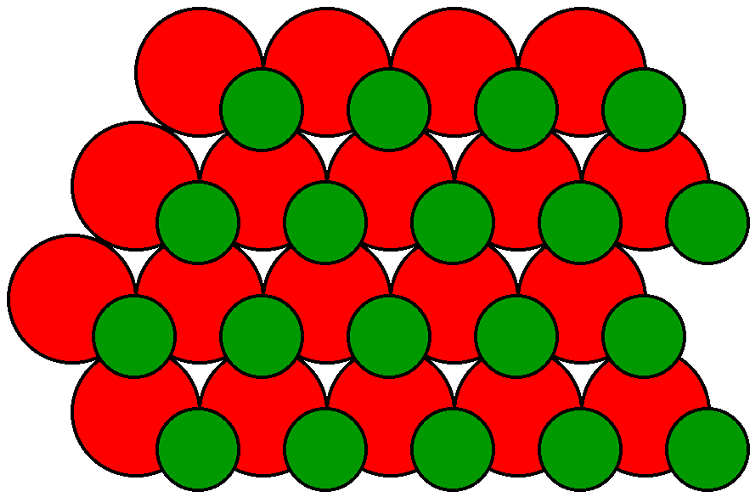
El (111) As es distinto del de (111) Ga debido a la diferencia en el número de electrones en la superficie. Como consecuencia, la (111) As cara graba más rápidamente que la cara (111) Ga. Además, la evaporación superficial por debajo de 770 °C ocurre más rápidamente en la cara (111) As.