7.3: Cristalografía de rayos X
- Page ID
- 70954

Introducción a la difracción de rayos X
Historia de la Cristalografía de Rayos X
El nacimiento de la cristalografía de rayos X es considerado por muchos como marcado por la formulación de la ley de ángulos constantes por Nicolaus Steno en 1669 (Figura\(\PageIndex{1}\)).
Si bien Steno es bien conocido por sus numerosos principios respecto a todas las áreas de la vida, esta ley particular que trata de formas geométricas y celosías cristalinas es un terreno familiar para todos los químicos. Simplemente afirma que los ángulos entre las caras correspondientes sobre los cristales son los mismos para todos los especímenes del mismo mineral. La importancia de esto para la química es que dado este hecho, los sólidos cristalinos serán fácilmente identificables una vez que se haya establecido una base de datos. Al igual que resolver un rompecabezas, las estructuras cristalinas de compuestos heterogéneos podrían resolverse de manera muy metódica comparando la composición química y sus interacciones.

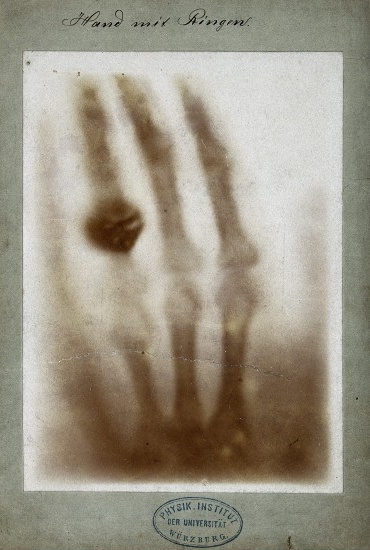
Si bien a Steno se le dio crédito por la noción de cristalografía, el hombre que proporcionó las herramientas necesarias para llevar la cristalografía a la arena científica fue Wilhelm Roentgen (Figura\(\PageIndex{2}\)), quien en 1895 fue pionero con éxito en una nueva forma de fotografía, una que supuestamente podría penetrar a través del papel, madera y carne humana; debido al desconocimiento del funcionamiento específico de este nuevo descubrimiento, la comunidad científica etiquetó convenientemente las nuevas partículas radiografías. Este evento desencadenó una reacción en cadena de experimentos y estudios, no todos realizados por físicos. En un solo mes, los médicos estaban usando rayos X para identificar objetos extraños como en el cuerpo humano como balas y cálculos renales (Figura\(\PageIndex{3}\)).

El crédito por el descubrimiento real de la difracción de rayos X es para Max von Laue (Figura\(\PageIndex{4}\), a quien se le otorgó el Premio Nobel de Física en 1914 por el descubrimiento de la difracción de rayos X. Cuenta la leyenda que la noción que finalmente llevó a un premio Nobel nació en un jardín de Munich, mientras que von Laue reflexionaba sobre el problema de pasar ondas de radiación electromagnética a través de una disposición cristalina específica de átomos. Debido a la longitud de onda relativamente grande de la luz visible, von Laue se vio obligado a dirigir su atención a otra parte del espectro electromagnético, hacia donde residieron longitudes de onda más cortas. Apenas unas décadas antes, Röentgen había anunciado públicamente el descubrimiento de los rayos X, que supuestamente tenían una longitud de onda más corta que la de la luz visible. Al contar con esta información, von Laue confió la tarea de realizar el trabajo experimental a dos técnicos, Walter Friedrich y Paul Knipping. La instalación consistió en una fuente de rayos X, la cual transmitió radiación directamente a un cristal de sulfato de cobre alojado en una caja de plomo. La película se alineó contra los lados y la parte posterior de la caja, para capturar el haz de rayos X y su patrón de difracción. El desarrollo de la película mostró un círculo oscuro en el centro de la película, rodeado por varios círculos extremadamente bien definidos, los cuales se habían formado como resultado de la difracción del haz de rayos X por la disposición geométrica ordenada del sulfato de cobre. Max von Laue procedió entonces a elaborar las fórmulas matemáticas involucradas en el patrón de difracción observado, por lo que fue galardonado con el Premio Nobel de Física en 1914.

Principios de la Difracción de Rayos X (XRD)
La definición más simple de difracción son las irregularidades causadas cuando las ondas se encuentran con un objeto. La difracción es un fenómeno que existe comúnmente en las actividades cotidianas, pero que a menudo se ignora y se da por sentado. Por ejemplo, al mirar el lado de información de un disco compacto, a menudo aparecerá un patrón de arcoíris cuando capta luz en cierto ángulo. Esto es causado por la luz visible que golpea los surcos del disco, produciendo así un efecto arcoíris (Figura\(\PageIndex{5}\)), tal como lo interpretan los ojos de los observadores. Otro ejemplo es la formación de anillos aparentemente concéntricos alrededor de un objeto astronómico de luminosidad significativa cuando se observa a través de las nubes. Las partículas que componen las nubes difractan la luz del objeto astronómico alrededor de sus bordes, provocando la ilusión de anillos de luz alrededor de la fuente. Es fácil olvidar que la difracción es un fenómeno que se aplica a todas las formas de ondas, no solo a la radiación electromagnética. Debido a la gran variedad de posibles tipos de difracciones, se han acuñado muchos términos para diferenciar entre tipos específicos. El tipo de difracción más prevalente a la cristalografía de rayos X se conoce como difracción de Bragg, la cual se define como la dispersión de ondas desde una estructura cristalina.

Formulada por William Lawrence Bragg (Figura\(\PageIndex{6}\)), la ecuación de la ley de Bragg relaciona la longitud de onda con el ángulo de incidencia y el espaciado de celosía,\ ref {1}, donde n es una constante numérica conocida como el orden del haz difractado, λ es la longitud de onda del haz, d denota la distancia entre planos de celosía, y θ representa el ángulo de la onda difractada. Las condiciones dadas por esta ecuación deben cumplirse si se va a producir difracción.
\[ n\lambda \ =\ 2d\ sin(\theta ) \label{1} \]

Debido a la naturaleza de la difracción, las ondas experimentarán interferencia constructiva (Figura\(\PageIndex{7}\)) o destructiva (Figura\(\PageIndex{8}\)) con otras ondas. De la misma manera, cuando un haz de rayos X se difracta de un cristal, las diferentes partes del haz difractado tendrán energía aparentemente más fuerte, mientras que otras partes habrán parecido haber perdido energía. Esto depende principalmente de la longitud de onda del haz incidente y del espaciamiento entre las celosías cristalinas de la muestra. La información sobre la estructura reticular se obtiene variando las longitudes de onda del haz, los ángulos incidentes y la orientación del cristal. Al igual que resolver un rompecabezas, se puede construir una estructura tridimensional del sólido cristalino observando cambios en los datos con variación de las variables mencionadas anteriormente.
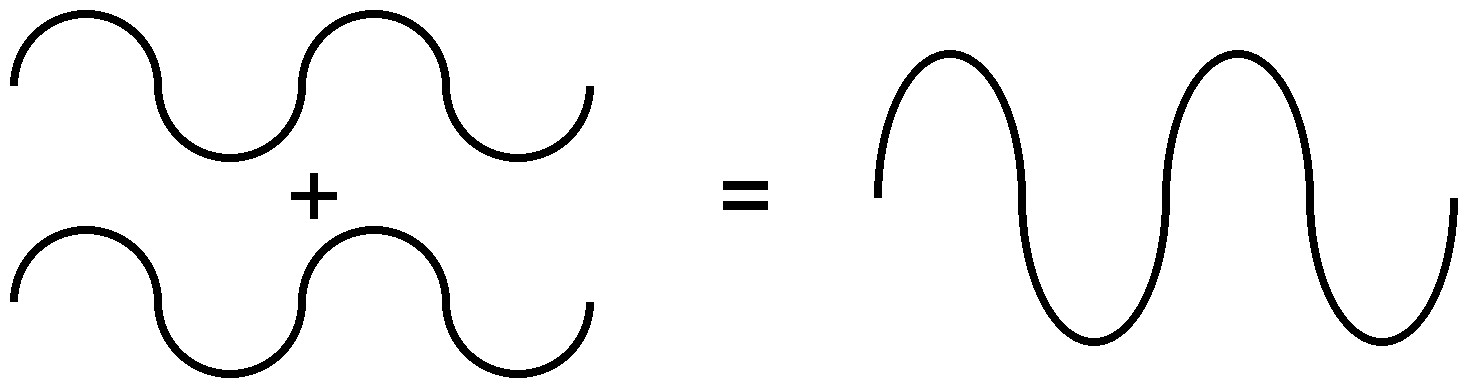
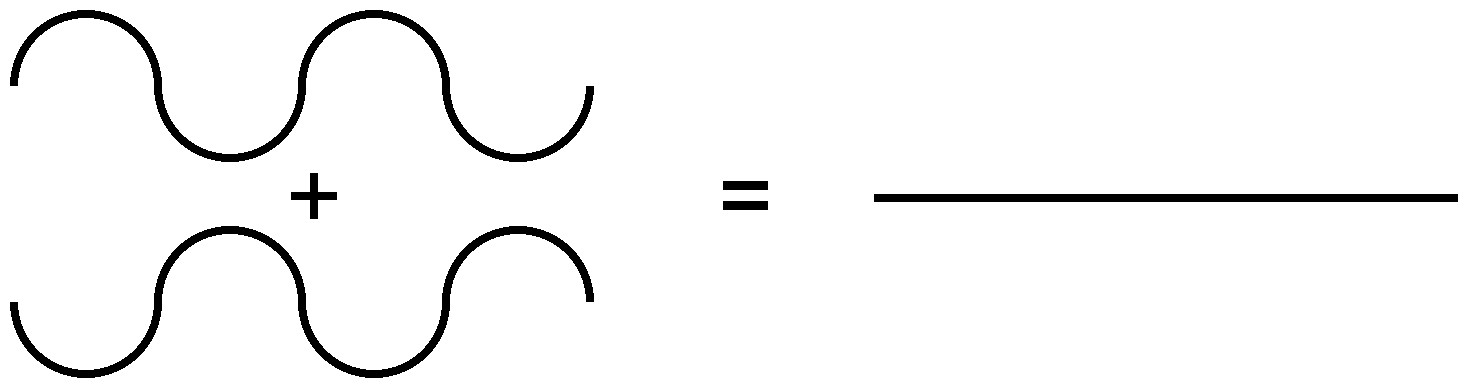
El difractómetro de rayos X
En el corazón de cualquier máquina XRD se encuentra la fuente de rayos X. Las máquinas modernas generalmente confían en el metal cobre como el elemento de elección para producir rayos X, aunque hay variaciones entre los diferentes fabricantes. Debido a que los patrones de difracción se registran durante un período prolongado de tiempo durante el análisis de la muestra, es muy importante que la intensidad del haz permanezca constante durante todo el análisis, o de lo contrario se obtendrán datos defectuosos. Ante esto, incluso antes de que se genere un haz de rayos X, la corriente debe pasar a través de un voltaje regular, lo que garantizará un flujo constante de voltaje a la fuente de rayos X.
Otro componente crucial para el análisis de los cristalinos vía rayos X es el detector. Cuando se desarrolló por primera vez la XRD, la película fue el método más utilizado para reconocer patrones de difracción. La desventaja más obvia de usar película es el hecho de que tiene que reemplazarse cada vez que se introduce un nuevo espécimen, lo que hace que la recolección de datos sea un proceso que consume tiempo. Además, la película solo se puede usar una vez, lo que lleva a un aumento en el costo del análisis de difracción de operación.
Desde los orígenes de XRD, los métodos de detección han progresado hasta el punto en que las modernas máquinas XRD están equipadas con detectores de semiconductores, que producen pulsos proporcionales a la energía absorbida. Con estos modernos detectores, hay dos formas generales en las que se puede obtener un patrón de difracción. El primero se llama escaneo continuo, y es exactamente lo que implica el nombre. El detector se pone en un movimiento circular alrededor de la muestra, mientras que un haz de rayos X se dispara constantemente en la muestra. Los pulsos de energía se representan con respecto al ángulo de difracción, lo que asegura que se registren todos los rayos X difractados. El segundo y más ampliamente utilizado método se conoce como escaneo por pasos. El escaneo escalonado tiene similitud con el escaneo continuo, excepto que es altamente computarizado y mucho más eficiente. En lugar de mover el detector en un círculo alrededor de toda la muestra, el escaneo escalonado implica recopilar datos en un ángulo fijo a la vez, de ahí el nombre. Dentro de estos parámetros de detección, los tipos de detectores pueden ser variados por sí mismos. Un tipo más común de detector, conocido como el detector de dispositivo de carga acoplada (CCD) (Figura\(\PageIndex{9}\), se puede encontrar en muchas máquinas XRD, debido a su rápida capacidad de recolección de datos. Un detector CCD está compuesto por numerosas rejillas sensibles a la radiación, cada una conectada a sensores que miden los cambios en la radiación electromagnética. Otro tipo de detector comúnmente visto es un simple contador de centelleo (Figura\(\PageIndex{10}\)), que cuenta la intensidad de los rayos X que encuentra a medida que se mueve a lo largo de un eje de rotación. Una analogía comparable a las diferencias entre los dos detectores mencionados sería que el detector CCD es capaz de ver en dos dimensiones, mientras que los contadores de centelleo solo son capaces de ver en una dimensión.


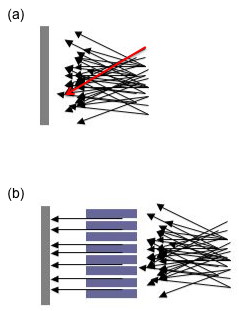
Aparte de los dos componentes anteriores, hay muchas otras variables involucradas en el análisis de muestras por una máquina XRD. Como se mencionó anteriormente, un haz incidente estable es extremadamente importante para una buena recolección de datos. Para asegurar aún más esto, a menudo habrá lo que se conoce como una hendidura Söller o colimador que se encuentra en muchas máquinas XRD. Una hendidura de Söller colisiona la dirección del haz de rayos X. En el haz de rayos X colimado los rayos son paralelos, y por lo tanto se extenderán mínimamente a medida que se propaguen (Figura\(\PageIndex{11}\). Sin un colimador se registrarán rayos X desde todas las direcciones; por ejemplo, un rayo que haya pasado por la parte superior del espécimen (ver la flecha roja en la Figura\(\PageIndex{11}\) a) pero que pasa a estar viajando en dirección descendente puede ser registrado en la parte inferior de la placa. La imagen resultante será tan borrosa e indistinta que será inútil. Algunas máquinas tienen una hendidura Söller entre la muestra y el detector, lo que reduce drásticamente la cantidad de ruido de fondo, especialmente cuando se analizan muestras de hierro con una fuente de rayos X de cobre.
Esta máquina XRD monocristal (Figura\(\PageIndex{12}\)) cuenta con una línea de gas de enfriamiento, que permite al usuario bajar la temperatura de una muestra considerablemente por debajo de la temperatura ambiente. Hacerlo permite oportunidades para estudios realizados donde la muestra se mantiene en un estado de energía extremadamente baja, negando mucho movimiento vibracional que podría interferir con la recolección de datos consistentes de patrones de difracción. Además, se puede recopilar información sobre los efectos de la temperatura en una estructura cristalina. También se ve en la Figura\(\PageIndex{13}\) el objeto en forma de gancho ubicado entre el emisor de haz y el detector. Sirve para bloquear los rayos X que no fueron difractados de ser vistos por el detector, reduciendo drásticamente la cantidad de ruido innecesario que de otro modo oscurecería el análisis de datos.
Evolución de XRD en Polvo
Con el tiempo, el análisis XRD ha evolucionado de un campo muy estrecho y específico a algo que abarca una rama mucho más amplia del ámbito científico. En sus primeras etapas, la XRD se limitó (a excepción de las estructuras más simples) al análisis monocristalino, ya que los métodos de detección no habían avanzado hasta un punto en el que se pudieran realizar procedimientos más complicados. Después de muchos años de descubrimiento y refinación, sin embargo, la tecnología ha progresado hasta donde las propiedades cristalinas (estructura) de los sólidos se pueden extraer directamente de una muestra de polvo, ofreciendo así información para muestras que no se pueden obtener como un solo cristal. Un área en la que esto es particularmente útil son los farmacéuticos, ya que muchos de los compuestos estudiados no están disponibles en forma monocristalina, solo en polvo.
Aunque la difracción monocristalina y la difracción de polvo esencialmente generan los mismos datos, debido a la naturaleza en polvo de esta última muestra, las líneas de difracción a menudo se superponen e interfieren con la recolección de datos. Esto es aparentemente especialmente cuando el ángulo de difracción 2 θ es alto; los patrones que emergen serán casi hasta el punto de no identificables, debido a la interrupción de los patrones de difracción individuales. Por esta razón particular, se ha creado un nuevo enfoque para interpretar los datos de difracción de polvo.
Existen dos métodos principales para interpretar los datos de difracción:
- El primero se conoce como el método tradicional, que es muy sencillo, y se parece al análisis de datos monocristalinos. Este método implica un proceso de dos etapas: 1) se recogen las intensidades y patrones de difracción de la muestra, y 2) se analizan los datos para producir una estructura cristalina. Sin embargo, como se mencionó anteriormente, los datos de una muestra en polvo a menudo están oscurecidos por múltiples patrones de difracción, lo que disminuye la probabilidad de que la estructura generada sea correcta.
- El segundo método se llama enfoque de espacio directo. Este método aprovecha que con la tecnología actual, los datos de difracción pueden calcularse para cualquier molécula, sea o no la molécula en cuestión. Incluso antes de que se recopilen los datos de difracción reales, se genera una gran cantidad de patrones teóricos de moléculas sospechosas por computadora, y se comparan con los datos experimentales. Con base en la correlación y en qué medida el patrón teórico se ajusta mejor a los datos experimentales, se formula una suposición sobre qué compuesto se cuestiona. Este método ha sido dado un paso más allá para imitar las interacciones sociales en una comunidad. Por ejemplo, se permite que las moléculas de ensayo teóricas de primera generación, después de compararlas con los datos experimentales, evolucionen dentro de los parámetros establecidos por los investigadores. Además, si procede, las moléculas se producen descendencia con otras moléculas, dando lugar a una segunda generación de moléculas, que se ajustan aún mejor a los datos experimentales. Al igual que un entorno natural, las mutaciones genéticas y la selección natural se introducen en la imagen, dando lugar en última instancia a una estructura molecular que representa los datos recopilados del análisis XRD.
Otro aspecto importante de poder estudiar compuestos en forma de polvo para el investigador farmacéutico es la capacidad de identificar estructuras en su estado natural. Una gran mayoría de los medicamentos en esta época se entregan a través de forma de polvo, ya sea en forma de píldora o cápsula. Los procesos de cristalización a menudo pueden alterar la composición química de la molécula (por ejemplo, mediante la inclusión de moléculas de disolvente) y, por lo tanto, estropear los datos si se limitan al análisis monocristalino. Además, cuando la muestra está en forma de polvo, existen otras variables que se pueden ajustar para ver los efectos en tiempo real sobre la molécula. La temperatura, la presión y la humedad son factores que se pueden cambiar in situ para obtener datos sobre cómo un medicamento podría responder a los cambios en esas variables particulares.
Difracción de Rayos X
Introducción
La difracción de rayos X en polvo (XRD) fue desarrollada en 1916 por Debye (Figura\(\PageIndex{12}\)) y Scherrer (Figura\(\PageIndex{13}\)) como una técnica que podría aplicarse donde no se puede realizar la difracción monocristalina tradicional. Esto incluye los casos en los que la muestra no se puede preparar como un solo cristal de tamaño y calidad suficientes. Las muestras de polvo son más fáciles de preparar y son especialmente útiles para la investigación farmacéutica.

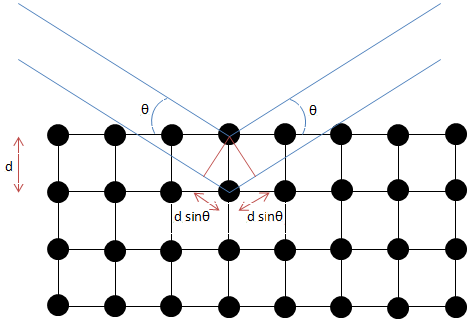
La difracción ocurre cuando una onda se encuentra con un conjunto de objetos de dispersión regularmente espaciados, y su longitud de onda de la distancia entre los objetos de dispersión es del mismo orden de magnitud. Esto hace que los rayos X sean adecuados para la cristalografía, ya que sus parámetros de longitud de onda y celosía cristalina están ambos en la escala de angstroms (Å). La difracción cristalina se puede describir por difracción de Bragg,\ ref {2}, donde λ es la longitud de onda de la radiografía monocromática incidente, d es la distancia entre planos cristalinos paralelos, y θ el ángulo entre el haz y el plano.
\[ \lambda \ =\ 2d\ sin \theta \label{2} \]
Para que ocurra interferencia constructiva entre dos ondas, la diferencia de longitud de trayectoria entre las ondas debe ser un múltiplo integral de su longitud de onda. Esta diferencia de longitud de trayectoria está representada por 2d sinθ Figura\(\PageIndex{14}\). Debido a que el sinθ no puede ser mayor que 1, la longitud de onda de los rayos X limita el número de picos de difracción que pueden aparecer.
Producción y Detección de Rayos X
La mayoría de los difractómetros utilizan Cu o Mo como fuente de rayos X, y específicamente la radiación K α de longitudes de onda de 1.54059 Å y 0.70932 Å, respectivamente. Una corriente de electrones se acelera hacia el ánodo objetivo metálico desde un cátodo de tungsteno, con una diferencia de potencial de aproximadamente 30-50 kV. Como esto genera mucho calor, el ánodo objetivo debe enfriarse para evitar que se derrita.
La detección del haz difractado se puede hacer de muchas maneras, y un sistema común es el contador proporcional de gas (GPC). El detector se llena con un gas inerte como el argón, y se crean pares electrón-ion cuando los rayos X pasan a través de él. Una diferencia de potencial aplicada separa los pares y genera ionizaciones secundarias a través de un efecto de avalancha. La amplificación de la señal es necesaria ya que la intensidad del haz difractado es muy baja en comparación con el haz incidente. La corriente detectada es entonces proporcional a la intensidad del haz difractado. Un GPC tiene un fondo de muy bajo ruido, lo que lo hace ampliamente utilizado en laboratorios.
Realizar difracción de rayos X
La exposición a rayos X puede tener consecuencias para la salud, siga los procedimientos de seguridad al usar el difractómetro.
La distribución del tamaño de partícula debe ser uniforme para asegurar que el patrón de difracción no esté dominado por unas pocas partículas grandes cerca de la superficie. Esto se puede hacer moliendo la muestra para reducir el tamaño promedio de partícula a <10µm. Sin embargo, si los tamaños de partícula son demasiado pequeños, esto puede conducir a la ampliación de los picos. Esto se debe tanto al daño de la celosía como a la reducción del número de planos que provocan interferencias destructivas.
El patrón de difracción se compone en realidad de ángulos que no sufrieron interferencia destructiva debido a su relación especial descrita por la Ley de Bragg (Figura\(\PageIndex{15}\)). Si la interferencia destructiva se reduce cerca de estos ángulos especiales, el pico se ensancha y se vuelve menos distinto. Algunos cristales como la calcita (CaCo 3, Figura\(\PageIndex{15}\) tienen orientaciones preferidas y cambiarán su orientación cuando se aplique presión. Esto conduce a diferencias en el patrón de difracción de muestras 'sueltas' y prensadas. Por lo tanto, es importante evitar incluso tocar polvos 'sueltos' para evitar errores a la hora de recolectar datos.
El polvo de muestra se carga en una placa de muestra para su montaje en el difractómetro (Figura\(\PageIndex{16}\)), donde los brazos giratorios que contienen la fuente de rayos X y el detector escanean la muestra en diferentes ángulos de incidencia. La placa de muestra se gira horizontalmente durante el escaneo para asegurar que el polvo se exponga uniformemente a los rayos X.

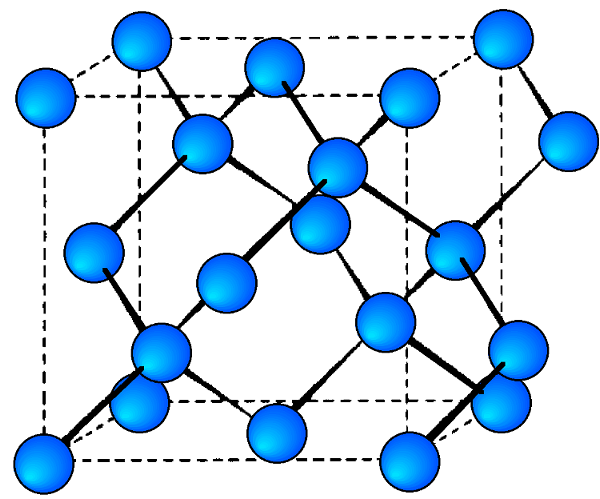
En la Figura se muestra un espectro de difracción de rayos X de muestra de germanio\(\PageIndex{17}\), con picos identificados por los planos que causaron esa difracción. El germanio tiene una red cristalina cúbica de diamante (Figura\(\PageIndex{18}\)), que lleva el nombre de la estructura cristalina de ejemplo prototípico. La estructura cristalina determina qué planos cristalinos causan la difracción y los ángulos en los que ocurren. Los ángulos se muestran en 2θ ya que es el ángulo medido entre los dos brazos del difractómetro, es decir, el ángulo entre el haz incidente y el haz difractado (Figura\(\PageIndex{14}\)).
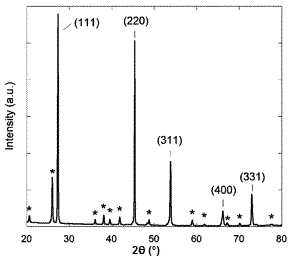
Determinación de la estructura cristalina para celosías cúbicas
Hay tres celosías básicas de cristal cúbico, y son el cúbico simple (SC), el cúbico centrado en el cuerpo (BCC) y la figura cúbica centrada en la cara (FCC)\(\PageIndex{19}\). Estas estructuras son lo suficientemente simples como para tener sus espectros de difracción analizados sin la ayuda de software.
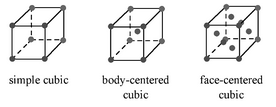
Cada una de estas estructuras tiene reglas específicas sobre cuáles de sus planos pueden producir difracción, basadas en sus índices Miller (hkl).
- Las redes SC muestran difracción para todos los valores de (hkl), por ejemplo, (100), (110), (111), etc.
- Las redes BCC muestran difracción cuando la suma de h + k + l es par, e.g., (110), (200), (211), etc.
- Las redes FCC muestran difracción cuando los valores de (hkl) son todos pares o todos impares, por ejemplo, (111), (200), (220), etc.
- Las redes cúbicas de diamante como la del germanio son estructuras FCC con cuatro átomos adicionales en las esquinas opuestas de los intersticios tetraédricos. Muestran difracción cuando los valores de (hkl) son todos impares o todos pares y la suma h + k + l es un múltiplo de 4, e.g., (111), (220), (311), etc.
El orden en que aparecen estos picos depende de la suma de h 2 + k 2 + l 2. Estos se muestran en la Tabla\(\PageIndex{1}\).
| (hkl) | h 2 +k 2 +l 2 | BCC | FCC |
| 100 | 1 | ||
| 110 | 2 | Y | |
| 111 | 3 | Y | |
| 200 | 4 | Y | Y |
| 210 | 5 | ||
| 211 | 6 | Y | |
| 220 | 8 | Y | Y |
| 300, 221 | 9 | ||
| 310 | 10 | Y | |
| 311 | 11 | Y | |
| 222 | 12 | Y | Y |
| 320 | 13 | ||
| 321 | 14 | Y | |
| 400 | 16 | Y | Y |
| 410, 322 | 17 | ||
| 411, 330 | 18 | Y | |
| 331 | 19 | Y | |
| 420 | 20 | Y | Y |
| 421 | 21 |
El valor de d para cada uno de estos planos se puede calcular usando\ ref {3}, donde a es el parámetro de celosía del cristal.
La constante de celosía, o parámetro de celosía, se refiere a la distancia constante entre celdas unitarias en una red cristalina.
\[ \frac{1}{d^{2}} \ =\ \frac{h^{2}+k^{2}+l^{2}}{a^{2}} \label{3} \]
Como la estructura cúbica de diamante de Ge puede ser complicada, a continuación se muestra un ejemplo trabajado más simple para la difracción de muestras de NaCl con radiación Cu-K α. Dados los valores de 2θ que resultan en difracción, se\(\PageIndex{2}\) puede construir Tabla.
| 2θ | θ | Sinθ | Sin 2 θ |
| 27.36 | 13.68 | 0.24 | 0.0559 |
| 31.69 | 15.85 | 0.27 | 0.0746 |
| 45.43 | 22.72 | 0.39 | 0.1491 |
| 53.85 | 26.92 | 0.45 | 0.2050 |
| 56.45 | 28.23 | 0.47 | 0.2237 |
| 66.20 | 33.10 | 0.55 | 0.2982 |
| 73.04 | 36.52 | 0.60 | 0.3541 |
| 75.26 | 37.63 | 0.61 | 0.3728 |
Los valores de estas relaciones pueden entonces ser inspeccionados para ver si corresponden a una serie esperada de valores hkl. En este caso, la última columna da una lista de números enteros, que corresponde a los valores h 2 + k 2 + l 2 de la difracción reticular FCC. De ahí que el NaCl tenga una estructura FCC, mostrada en ángulos Figura\(\PageIndex{20}\).
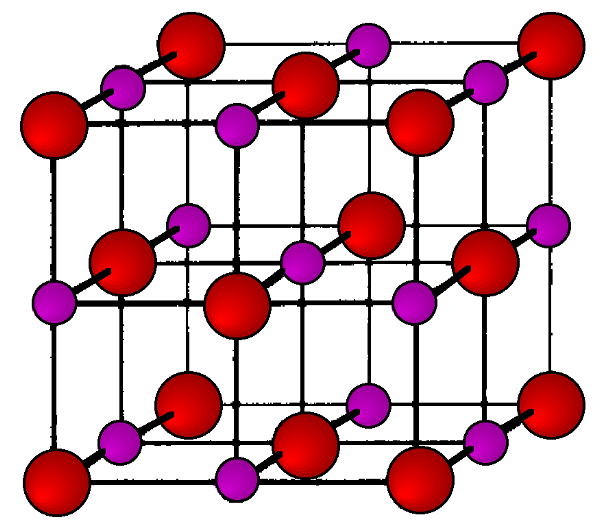
El parámetro reticular de NaCl ahora se puede calcular a partir de estos datos. El primer pico ocurre a θ = 13.68°. Dado que la longitud de onda de la radiación Cu-K α es 1.54059 Å, la Ecuación de Bragg\ ref {4} se puede aplicar de la siguiente manera:
\[ 1.54059 \ =\ 2d\ sin 13.68 \label{4} \]
\[ d\ =\ 3.2571\ Å \label{5} \]
Dado que el primer pico corresponde al plano (111), la distancia entre dos planos paralelos (111) es de 3.2571 Å. El parámetro de celosía ahora se puede elaborar usando\ ref {6}.
\[ 1/3.2561^{2}\ =\ (1^{2}+1^{2}+I^{2})/a^{2} \label{6} \]
\[ a\ =\ 5.6414\ Å \label{7} \]
El espectro de XRD en polvo de nanopartículas de Ag se da en la Figura\(\PageIndex{21}\) como recolectado usando radiación Cu-K α de 1.54059 Å. Determinar su estructura cristalina y el parámetro de celosía usando los picos marcados.
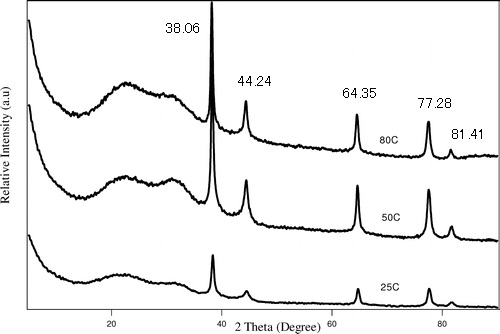
| 2θ | θ | Sinθ | Sin 2 θ | Sin 2 θ/SIN 2 θ | 2 x Sin 2 θ/Sin 2 θ | 3 x Sin 2 θ/Sin 2 θ |
| 38.06 | 19.03 | 0.33 | 0.1063 | 1.00 | 2.00 | 3.00 |
| 44.24 | 22.12 | 0.38 | 0.1418 | 1.33 | 2.67 | 4.00 |
| 64.35 | 32.17 | 0.53 | 0.2835 | 2.67 | 5.33 | 8 |
| 77.28 | 38.64 | 0.62 | 0.3899 | 3.67 | 7.34 | 11 |
| 81.41 | 40.71 | 0.65 | 0.4253 | 4 | 8 | 12 |
| 97.71 | 48.86 | 0.75 | 0.5671 | 5.33 | 10.67 | 16 |
| 110.29 | 55.15 | 0.82 | 0.6734 | 6.34 | 12.67 | 19.01 |
| 114.69 | 57.35 | 0.84 | 0.7089 | 6.67 | 13.34 | 20.01 |
Aplicando la Ecuación de Bragg\ ref {8},
\[ 1.54059\ =\ 2d\ sin\ 19.03 \label{8} \]
\[ d\ =\ 2.3624\ Å \label{9} \]
Calcular el parámetro de celosía usando\ ref {10},
\[ 1/2.3624^{2}\ =\ (1^{2}+1^{2}+I^{2})/a^{2} \label{10} \]
\[ a\ =\ 4.0918\ Å \label{11} \]
La última columna da una lista de números enteros, que corresponde a los valores h 2 + k 2 + l 2 de la difracción reticular FCC. De ahí que las nanopartículas de Ag tengan una estructura FCC.
Determinar la composición
Como se vio anteriormente, cada cristal dará un patrón de picos de difracción basado en su tipo de celosía y parámetro. Estos patrones de huellas digitales se compilan en bases de datos como la del Comité Conjunto de Difracción de Polvo Estándar (JCPDS). Así, los espectros XRD de las muestras pueden ser emparejados con los almacenados en la base de datos para determinar su composición de manera fácil y rápida.
Monitoreo de reacción en estado sólido
La XRD en polvo también es capaz de realizar análisis en reacciones de estado sólido como la transición de dióxido de titanio (TiO 2) anatasa a rutilo. Un difractómetro equipado con una cámara de muestra que se puede calentar puede tomar difractogramas a diferentes temperaturas para ver cómo progresa la reacción. Los espectros del cambio en los picos de difracción durante esta transición se muestran en la Figura\(\PageIndex{22}\)\(\PageIndex{23}\), Figura y Figura\(\PageIndex{24}\).
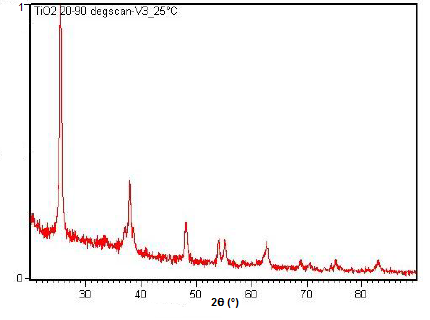
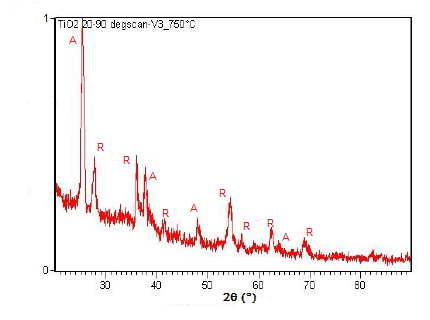
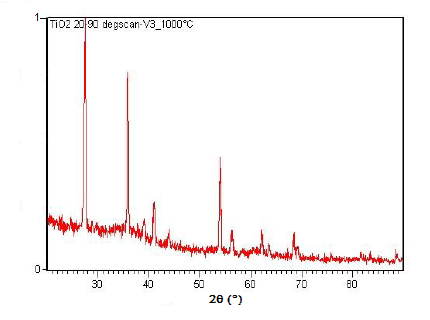
Resumen
La XRD permite una rápida determinación de la composición de muestras desconocidas y proporciona información sobre la estructura cristalina. La XRD en polvo es una aplicación útil de difracción de rayos X, debido a la facilidad de preparación de la muestra en comparación con la difracción monocristalina. Su aplicación al monitoreo de reacciones en estado sólido también puede proporcionar información sobre la estabilidad de fase y la transformación.
Introducción a la cristalografía de rayos X de cristal único
Descrito simplemente, la difracción de rayos X monocristalino (XRD) es una técnica en la que un cristal de una muestra en estudio es bombardeado con un haz de rayos X desde muchos ángulos diferentes, y se miden y registran los patrones de difracción resultantes. Al agregar los patrones de difracción y convertirlos a través de la transformada de Fourier en un mapa de densidad electrónica, se puede construir una celda unitaria que indica las posiciones atómicas promedio, las longitudes de enlace y las orientaciones relativas de las moléculas dentro del cristal.
Principios Fundamentales
Como analogía para describir los principios subyacentes de la difracción, imagínese brillar un láser sobre una pared a través de un tamiz fino. En lugar de observar un solo punto de luz en la pared, se observará un patrón de difracción, consistente en puntos de luz dispuestos regularmente, cada uno con una posición e intensidad definidas. El espaciamiento de estas manchas está inversamente relacionado con la rejilla en el tamiz: cuanto más fino es el tamiz, cuanto más separadas están las manchas, y cuanto más grueso es el tamiz, más cerca están las manchas. Los objetos individuales también pueden difractar la radiación si es de la longitud de onda apropiada, pero generalmente no se ve un patrón de difracción porque su intensidad es demasiado débil. La diferencia con un tamiz es que consiste en una rejilla hecha de alambres repetidos regularmente espaciados. Esta periodicidad magnifica en gran medida el efecto de difracción debido a la interferencia constructiva. A medida que los rayos de luz combinan amplitudes, la intensidad resultante de la luz que se ve en la pared es mucho mayor porque la intensidad es proporcional al cuadrado de la amplitud de la luz.
Para aplicar esta analogía a la XRD monocristalina, simplemente debemos escalarla hacia abajo. Ahora el tamiz es reemplazado por un cristal y el láser (luz visible) es reemplazado por un haz de rayos X. Aunque el cristal parece sólido y no en forma de rejilla, las moléculas o átomos contenidos dentro del cristal se disponen periódicamente, produciendo así el mismo efecto de aumento de intensidad que con el tamiz. Debido a que los rayos X tienen longitudes de onda que están en la misma escala que la distancia entre los átomos, pueden ser difractados por sus interacciones con la red cristalina.
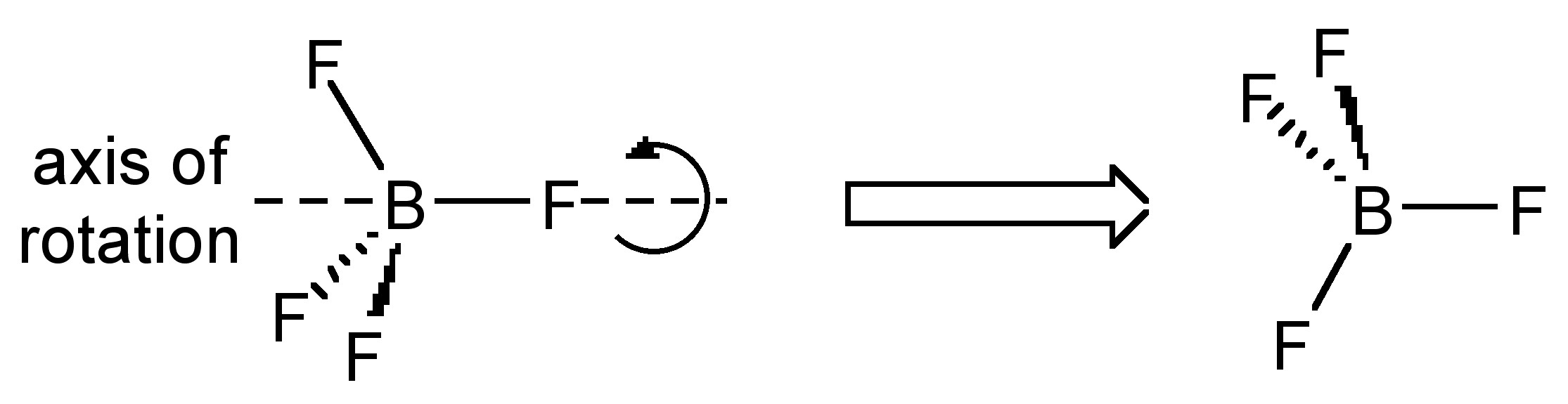
Estas interacciones son dictadas por la ley de Bragg, que dice que la interferencia constructiva ocurre solo cuando se satisface\ ref {12}; donde n es un entero, λ es la longitud de onda de la luz, d es la distancia entre planos paralelos en la red cristalina, y θ es el ángulo de incidencia entre el haz de rayos X y los planos de difracción (ver Figura\(\PageIndex{25}\)). Sin embargo, surge una complicación porque los cristales son periódicos en las tres dimensiones, mientras que el tamiz se repite en solo dos dimensiones. Como resultado, los cristales tienen muchos planos de difracción diferentes que se extienden en ciertas orientaciones basadas en el grupo de simetría del cristal. Por esta razón, es necesario observar patrones de difracción desde muchos ángulos y orientaciones diferentes del cristal para obtener una imagen completa de la red recíproca.
La red recíproca de una celosía (celosía de Bravais) es la celosía en la que se representa la transformada de Fourier de la funciónondulada espacial de la celosía original (o celosía directa). La celosía recíproca de una celosía recíproca es la celosía original.
\[ n \lambda \ =\ 2d\ sin \theta \label{12} \]
La red recíproca está relacionada con la red cristalina así como el tamiz se relaciona con el patrón de difracción: son inversas entre sí. Cada punto en el espacio real tiene un punto correspondiente en el espacio recíproco y están relacionados por 1/d; es decir, cualquier vector en el espacio real multiplicado por su vector correspondiente en el espacio recíproco da un producto de unidad. Los ángulos entre los pares correspondientes de vectores permanecen inalterados.
El espacio real es el dominio del cristal físico, es decir, incluye la red cristalina formada por los átomos físicos dentro del cristal. El espacio recíproco es, en pocas palabras, la transformada de Fourier del espacio real; prácticamente, vemos que los patrones de difracción resultantes de diferentes orientaciones del cristal de muestra en el haz de rayos X son en realidad proyecciones bidimensionales de la red recíproca. Así, al recolectar patrones de difracción de todas las orientaciones del cristal, es posible construir una versión tridimensional de la red recíproca y luego realizar una transformada de Fourier para modelar la red cristalina real.
Técnica
Difracción de monocristal versus polvo
Dos tipos comunes de difracción de rayos X son XRD en polvo y XRD monocristalino, los cuales tienen beneficios y limitaciones particulares. Si bien la XRD en polvo tiene una preparación de muestra mucho más simple, puede ser difícil obtener datos estructurales a partir de un polvo porque las moléculas de muestra están orientadas aleatoriamente en el espacio; sin la periodicidad de una red cristalina, la relación señal/ruido disminuye considerablemente y se vuelve difícil de separar reflexiones provenientes de las diferentes orientaciones de la molécula. La ventaja de la XRD en polvo es que se puede utilizar para identificar de manera rápida y precisa una sustancia conocida, o para verificar que dos muestras desconocidas sean del mismo material.
La XRD monocristalina es mucho más intensiva en tiempo y datos, pero en muchos campos es esencial para la determinación estructural de moléculas pequeñas y macromoléculas en estado sólido. Debido a la periodicidad inherente a los cristales, las pequeñas señales de las reflexiones individuales se magnifican a través de la interferencia constructiva. Esto se puede utilizar para determinar las posiciones espaciales exactas de los átomos en las moléculas y puede producir distancias de enlace e información conformacional. La dificultad de la XRD monocristalina es que los cristales individuales pueden ser difíciles de obtener y el instrumento en sí puede ser prohibitivo en cuanto a costos.
A continuación se muestra un ejemplo de patrones de difracción típicos para XRD monocristalino y en polvo ((Figura\(\PageIndex{27}\) y Figura\(\PageIndex{28}\), respectivamente). Los puntos de la primera imagen corresponden a las reflexiones de Bragg y juntos forman una sola visión del espacio recíproco de la molécula. En XRD en polvo, la orientación aleatoria de los cristales significa que las reflexiones de todos ellos se ven a la vez, produciendo los anillos de difracción observados que corresponden a vectores particulares en la red recíproca del material.
Técnica
En un experimento de difracción de rayos X monocristalino, el espacio recíproco de un cristal se construye midiendo los ángulos e intensidades de las reflexiones en los patrones de difracción observados. Estos datos se utilizan luego para crear un mapa de densidad electrónica de la molécula que se puede refinar para determinar las longitudes de enlace promedio y las posiciones de los átomos en el cristal.
Instrumentación
La configuración básica para XRD monocristalino consiste en una fuente de rayos X, un colimador para enfocar el haz, un goniómetro para sostener y rotar el cristal, y un detector para medir y registrar las reflexiones. Los instrumentos suelen contener un tope de haz para evitar que el haz de rayos X primario golpee el detector, y una cámara para ayudar a posicionar el cristal. Muchos también contienen una salida conectada a un suministro de gas frío (como nitrógeno líquido) con el fin de enfriar el cristal de muestra y reducir su movimiento vibratorio a medida que se recopilan los datos. Un instrumento típico se muestra en la Figura\(\PageIndex{28}\) y Figura\(\PageIndex{31}\).
Obtención de cristales individuales
A pesar de los avances en instrumentación y programas informáticos que hacen que la recolección de datos y la resolución de estructuras cristalinas sean significativamente más rápidas y fáciles, aún puede ser un desafío obtener cristales adecuados para el análisis. Los cristales ideales son simples, no hermanados, transparentes y de tamaño suficiente para montarse dentro del haz de rayos X (generalmente 0.1-0.3 mm en cada dirección). También tienen caras limpias y bordes lisos. A continuación se presentan imágenes de algunos cristales ideales (Figura\(\PageIndex{30}\) y Figura\(\PageIndex{31}\)), así como un ejemplo de cristales hermanados (Figura\(\PageIndex{32}\)).
El hermanamiento de cristales ocurre cuando dos o más cristales comparten puntos de celosía de manera simétrica. Esto generalmente da como resultado patrones de difracción complejos que son difíciles de analizar y construir una red recíproca.
La formación de cristales puede verse afectada por la temperatura, la presión, la elección del disolvente, la saturación, la nucleación y el sustrato. El crecimiento cristalino lento tiende a ser el mejor, ya que el crecimiento rápido crea más imperfecciones en la red cristalina e incluso puede conducir a un precipitado o gel. De manera similar, demasiados sitios de nucleación (puntos en los que comienza el crecimiento de los cristales) pueden conducir a muchos cristales pequeños en lugar de unos pocos, bien definidos.
Hay una serie de métodos básicos para cultivar cristales adecuados para XRD monocristalino:
- El método más básico es evaporar lentamente una solución saturada hasta que se sobresature y luego forma cristales. Esto a menudo funciona bien para el cultivo de cristales de moléculas pequeñas; las moléculas macroscópicas (como las proteínas) tienden a ser más difíciles.
- Una solución del compuesto a cristalizar se disuelve en un disolvente, luego un 'no disolvente' que es miscible con el primero pero en el que el propio compuesto es insoluble, se coloca cuidadosamente en capas encima de la solución. A medida que el no disolvente se mezcla con el disolvente por difusión, las moléculas de soluto son forzadas a salir de la solución y pueden formar cristales.
- Se coloca una solución cristalina en un pequeño recipiente abierto que luego se coloca en un recipiente cerrado más grande que contiene un no disolvente volátil. A medida que el no disolvente volátil se mezcla lentamente con la solución por difusión de vapor, el soluto se ve nuevamente obligado a salir de la solución, lo que a menudo conduce al crecimiento de cristales.
- Las tres técnicas anteriores se pueden combinar con siembra, donde un cristal del tipo deseado a cultivar se coloca en la solución saturada y actúa como sitio de nucleación y punto de partida para que comience el crecimiento cristalino. En algunos casos, esto puede incluso hacer que los cristales crezcan en una forma que normalmente no asumirían, ya que la semilla puede actuar como una plantilla que de otra manera no se seguiría.
- La técnica de gota colgante se usa típicamente para cultivar cristales de proteína. En esta técnica, se suspende una gota de solución concentrada de proteína (generalmente punteándola sobre un portaobjetos de microscopio recubierto de silicio) sobre un volumen mayor de la solución. Luego se sella todo el sistema y la lenta evaporación de la gota suspendida hace que se sobresature y forme cristales. (Una variación de esto es tener la gota de solución proteica descansando sobre una plataforma dentro del sistema cerrado en lugar de estar suspendida de la parte superior del recipiente).
Estas son sólo las formas más comunes en que se cultivan los cristales. Particularmente para macromoléculas, puede ser necesario probar cientos de condiciones de cristalización antes de obtener un cristal adecuado. Ahora existen técnicas automatizadas que utilizan robots para cultivar cristales, tanto para obtener grandes cantidades de cristales individuales como para realizar técnicas especializadas (como extraer un cristal de la solución) que de otro modo requerirían demasiado tiempo para ser de uso práctico.
Estudios de difracción de rayos X de gran angular de cristales líquidos
Algunas moléculas orgánicas muestran una serie de estados de transición intermedios entre los estados sólido e isotrópico líquido (Figura\(\PageIndex{33}\)) a medida que aumenta su temperatura. Estas fases intermedias tienen propiedades entre el sólido cristalino y el estado líquido isotrópico correspondiente, y por lo tanto se denominan fases cristalinas líquidas. Otro nombre es fases mesomórficas donde mesomórficas significa de forma intermedia. Según el físico de Gennes (Figura\(\PageIndex{34}\)), el cristal líquido es 'una fase intermedia, que tiene un orden similar al líquido en al menos una dirección y posee un grado de anisotropia'. Cabe señalar que todas las fases cristalinas líquidas están formadas por moléculas anisotrópicas (alargadas o en forma de disco) pero no todas las moléculas anisotrópicas forman fases cristalinas líquidas.
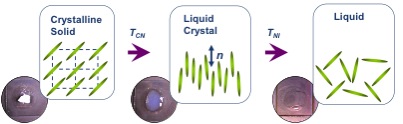

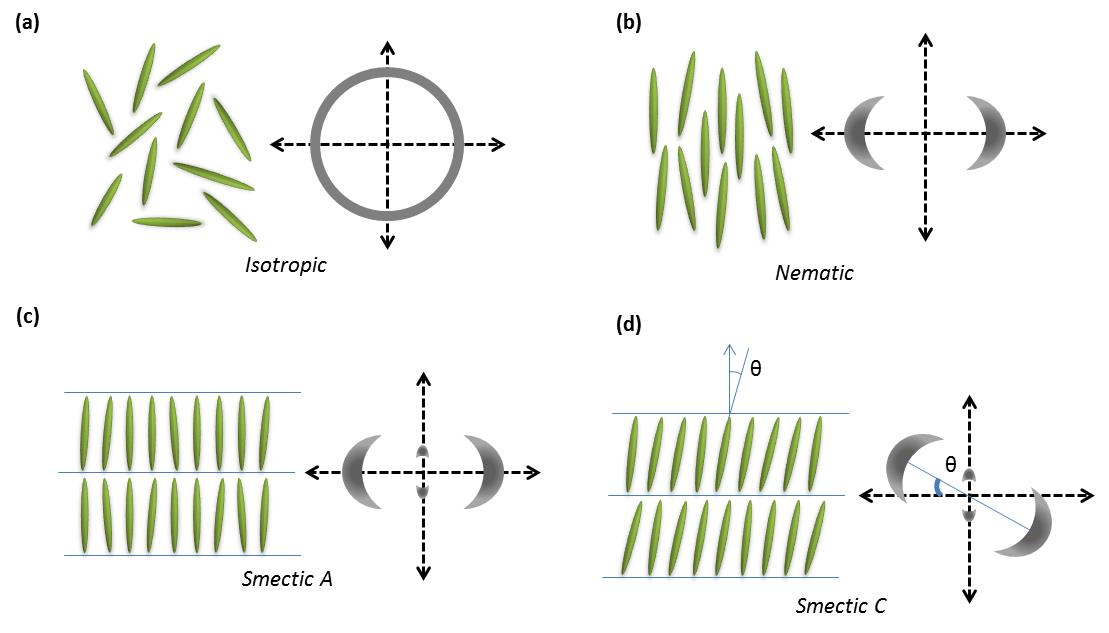
Los objetos anisotrópicos pueden poseer diferentes tipos de ordenamiento dando lugar a diferentes tipos de fases cristalinas líquidas (Figura\(\PageIndex{35}\)).
Fases Nemáticas
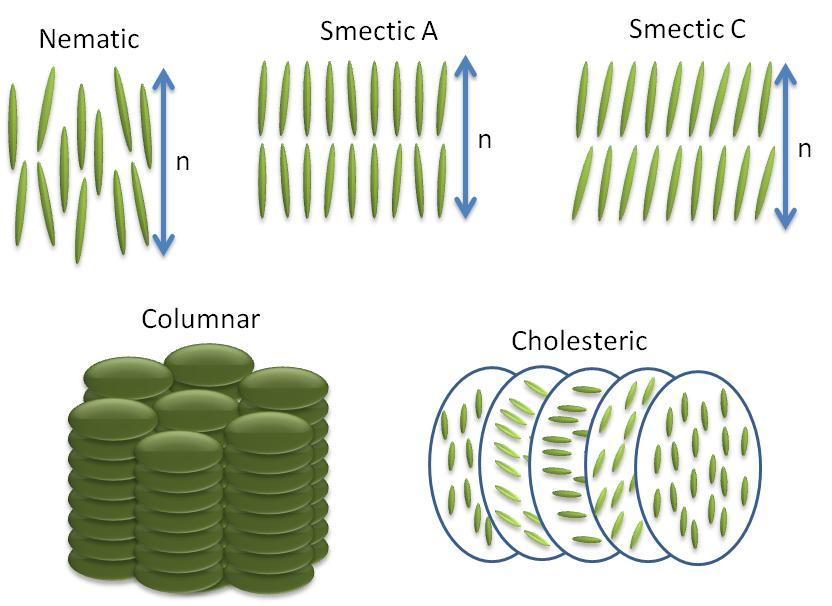
La palabra nemático proviene del griego para hilo, y se refiere a los defectos filiformes comúnmente observados en la microscopía óptica polarizante de estas moléculas. No tienen orden posicional solo orden orientacional, es decir, las moléculas todas pintas en la misma dirección. La dirección de las moléculas denotadas por el símbolo n comúnmente conocido como el 'director' (Figura\(\PageIndex{36}\)). El director n es bidireccional, lo que significa que los estados n y -n son indistinguibles.
Fases Esmeticas
Todas las fases esmécticas son estructuras estratificadas que suelen ocurrir a temperaturas ligeramente más bajas que las fases nemáticas. Hay muchas variaciones de las fases esmécticas, y algunas de las distintas son las siguientes:
- Cada capa en esméctica A es como un líquido bidimensional, y el eje largo de las moléculas es típicamente ortogonal a las capas (Figura\(\PageIndex{35}\).
- Al igual que los nemáticos, el estado n y -n son equivalentes. Están conformadas por moléculas aquirales y no polares.
- Al igual que con la esméctica A, la fase esméctica C está estratificada, pero el eje largo de las moléculas no es normal a lo largo de las capas. En cambio hace un ángulo (θ, Figura\(\PageIndex{35}\)). El ángulo de inclinación es un parámetro de orden de esta fase y puede variar de 0° a 45-50°.
- Las fases esmécticas C* son fases esmécticas formadas por moléculas quirales. Esta restricción añadida de quiralidad provoca una ligera distorsión de la estructura Esméctica C. Ahora la dirección de inclinación precede alrededor de la capa normal y forma una configuración helicoidal.
Fases colestericas
A veces, las fases colestéricas (Figura\(\PageIndex{35}\)) también se denominan fases nemáticas quirales porque son similares a las fases nemáticas en muchos aspectos. Muchos derivados del colesterol exhiben este tipo de fase. Generalmente están formados por moléculas quirales o dopando la matriz hospedadora nemática con moléculas quirales. Agregar quiralidad provoca distorsión helicoidal en el sistema, lo que hace que el director, n, gire continuamente en el espacio en forma de hélice con paso específico. La magnitud de la brea en una fase colestérica es una fuerte función de la temperatura.
Fases Columnares
En las fases columnares, las moléculas de cristales líquidos tienen la forma de discos en oposición a las de tipo varilla en las fases de cristal líquido nemático y esméctico. Estas moléculas en forma de disco se apilan en columnas y forman estructuras de matriz cristalina 2D (Figura\(\PageIndex{35}\)). Este tipo de ordenamiento bidimensional conduce a nuevas mesofases.
Introducción a la difracción de rayos X 2D
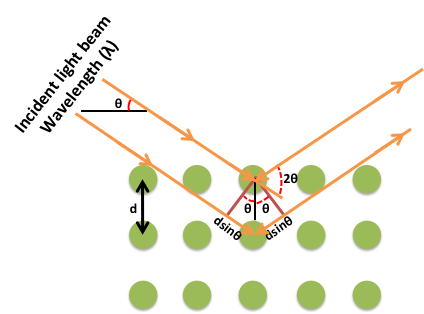
La difracción de rayos X (DRX) es una de las técnicas experimentales fundamentales utilizadas para analizar la disposición atómica de los materiales. El principio básico detrás de la difracción de rayos X es la Ley de Bragg (Figura\(\PageIndex{36}\)). Según esta ley, los rayos X que se reflejan desde los planos cristalinos adyacentes sufrirán interferencia constructiva solo cuando la diferencia de trayectoria entre ellos sea un múltiplo entero de la longitud de onda de los rayos X,\ ref {13}, donde n es un entero, d es el espaciamiento entre los planos cristalinos adyacentes, θ es el ángulo entre el haz de rayos X incidente y el plano de dispersión, y λ es la longitud de onda de los rayos X incidentes.
\[ 2d sin \theta \ =\ n \lambda \label{13}\ \]
Ahora la disposición atómica de las moléculas puede pasar de ser extremadamente ordenada (cristales individuales) a aleatoria (líquidos). Correspondientemente, los rayos X dispersos forman patrones de difracción específicos particulares de esa muestra. La figura\(\PageIndex{37}\) muestra la diferencia entre los rayos X dispersos de una muestra monocristalina y policristalina (polvo). En el caso de un solo cristal los rayos difractados apuntan a direcciones discretas (Figura\(\PageIndex{37}a\)), mientras que para la muestra policristalina los rayos difractados forman una serie de conos de difracción (Figura\(\PageIndex{37}b\)).

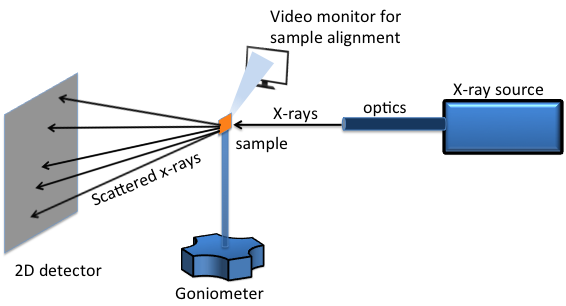
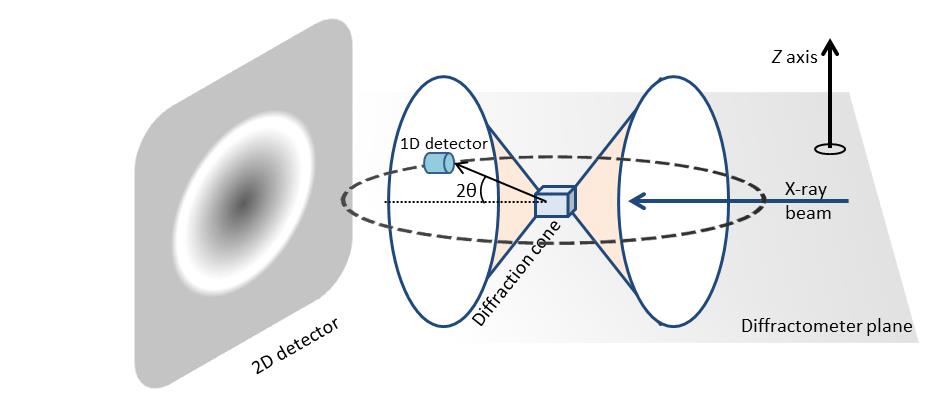
Un sistema de XRD bidimensional (2D) es un sistema de difracción con la capacidad de recolectar y analizar simultáneamente el patrón de difracción de rayos X en dos dimensiones. Una configuración típica de XRD 2D consta de cinco componentes principales (Figura\(\PageIndex{38}\)):
- Fuente de rayos X.
- Óptica de rayos X.
- Goniómetro.
- Dispositivo de alineación y monitoreo de muestras.
- Detector de área 2D.
Para los generadores de rayos X a escala de laboratorio, los rayos X se emiten bombardeando objetivos metálicos con electrones de alta velocidad acelerados por un campo eléctrico fuerte en el rango de 20-60 kV. Los diferentes objetivos metálicos que se pueden utilizar son el cromo (Cr), el cobalto (Co), el cobre (Cu), el molibdeno (Mo) y el hierro (Fe). Los más utilizados son Cu y Mo. Los sincrotrones son fuentes de radiación de energía aún mayor. Se pueden afinar para generar una longitud de onda específica y tienen una luminosidad mucho más brillante para una mejor resolución. Las instalaciones de sincrotrón disponibles en Estados Unidos son:
- Fuente de luz de radiación sincrotrón de Stanford (SSRL), Stanford, CA.
- Centro de Radiación Sincrotrón (SRC), Universidad de Wisconsin-Madison, Madison, WI.
- Fuente de luz avanzada (ALS), Lawrence Berkeley National, Berkeley, CA.
- Fuente Nacional de Luz Sincrotrón (NSLS), Laboratorio Nacional Brookhaven, Upton, NY.
- Fuente Avanzada de Fotones (APS), Laboratorio Nacional Argonne, Argonne, IL.
- Centro de Microestructuras Avanzadas y Dispositivos, Louisiana State University, Baton Rouge, LA.
- Fuente de Sincrotrón de Alta Energía de Cornell (CHEDREZ), Cornell, Ítaca, NY.
La óptica de rayos X está compuesta por el tubo de rayos X, monocromador, colimador estenopeico y tope del haz. Se utiliza un monocromador para eliminar la radiación de rayos X no deseada del tubo de rayos X. Se puede usar una difracción de un solo cristal para seleccionar una longitud de onda específica de radiación. Los materiales típicos utilizados son el grafito pirolítico y el silicio. Los haces de rayos X monocromáticos tienen tres componentes: rayos X paralelos, convergentes y divergentes. La función de un colimador estenopeico es filtrar el haz de rayos X incidente y permitir el paso de rayos X paralelos. Un detector de rayos X 2D puede ser una película o un detector digital, y su función es medir la intensidad de los rayos X difractados de una muestra en función de la posición, el tiempo y la energía.
Ventajas de 2D XRD en comparación con 1D XRD
Los datos de difracton 2D tienen mucha mas información en el patron de difraccion de comparacion, que se adquiere usando un detector 1D. La figura\(\PageIndex{39}\) muestra el patrón de difracción de una muestra policristalina. Solo con fines ilustrativos, se muestran dos conos de difracción en el esquema. En el caso de la difracción de rayos X 1D, el área de medición está confinada dentro de un plano etiquetado como plano difractómetro. El detector 1D se monta a lo largo del círculo de detección y no se considera la variación del patrón de difracción en la dirección z. El patrón de difracción recolectado es un promedio en un rango definido por un tamaño de haz en la dirección z. El patrón de difracción medido es una gráfica de la intensidad de rayos X en diferentes ángulos 2θ. Para la difracción de rayos X 2D, el área de medición no se limita al plano del difractómetro. En cambio, una gran parte de los anillos de difracción se miden simultáneamente dependiendo del tamaño del detector y la posición de la muestra.
Una de esas ventajas es la medición del porcentaje de cristalinidad de un material. Se requiere la determinación de la cristalinidad del material tanto para investigación como para control de calidad. La dispersión de materiales amorfos produce un anillo de intensidad difusa, mientras que las muestras policristalinas producen anillos o manchas afiladas y bien definidas. La capacidad de distinguir entre amorfo y cristalino es la clave para determinar con precisión el porcentaje de cristalinidad. Dado que la mayoría de las muestras cristalinas tienen orientación preferida, dependiendo de que la muestra esté orientada es posible medir diferentes picos o ningún pico usando un sistema de difracción convencional. Por otro lado, la orientación de la muestra no tiene efecto en la medición de difracción integrada de círculo completo realizada con detector 2D. Por lo tanto, una XRD 2D puede medir el porcentaje de cristalinidad con mayor precisión.
Patrones de difracción de rayos X de gran angular 2D de LC
Como se menciona en la sección de introducción, el cristal líquido es un estado intermedio entre las fases sólida y líquida. A temperaturas superiores a la temperatura de transición de fase de cristal líquido (Figura\(\PageIndex{40}\)), se convierten en líquido isotrópico, es decir, ausencia de orden posicional u orientacional de largo alcance dentro de las moléculas. Dado que un estado isotrópico no puede alinearse, su patrón de difracción consiste en anillos débiles y difusos Figura\(\PageIndex{40}a\). La razón por la que vemos algún patrón de difracción en el estado isotrópico es porque en los líquidos clásicos existe un orden posicional de corto alcance. El anillo tiene un radio de 4.5 Å y aparece mayormente a 20.5°. Representa la distancia entre las moléculas a lo largo de sus anchuras.
Las fases cristalinas líquidas nemáticas tienen orden orientacional de largo alcance pero no tienen orden posicional. Una muestra no alineada de cristal líquido nemático tiene un patrón de difracción similar al estado isotrópico. Pero en lugar de un anillo difuso, tiene una distribución de intensidad más nítida. Para una muestra alineada de cristal líquido nemático, los patrones de difracción de rayos X exhiben dos conjuntos de arcos difusos (Figura\(\PageIndex{40}\) b). El arco difuso en el radio mayor (P1, 4.5 Å) representa la distancia entre las moléculas a lo largo de sus anchuras. Bajo la presencia de un campo magnético externo, las muestras con anisotropía diamagnética positiva se alinean paralelas al campo y P1 se orienta perpendicularmente al campo. Mientras que las muestras con anisotropía diamagnética negativa se alinean perpendicularmente al campo con P1 siendo paralelo al campo. La distribución de intensidad dentro de estos arcos representa la extensión de alineación dentro de la muestra; generalmente denotada por S.
La anistropía diamagnética de todos los cristales líquidos con anillo aromático es positiva, y del orden de 10 -7. El valor disminuye con la sustitución de cada anillo aromático por un ciclohexano u otro grupo alifático. Se observa anistropía diamagnética negativa para LC puramente cicloalifáticas.
Cuando una fase esméctica se enfría lentamente bajo la presencia del campo externo, se observan dos conjuntos de picos difusos en patrón de difracción (Figura\(\PageIndex{40}\) c). El pico difuso en ángulos pequeños se condensa en picos agudos cuasi-Bragg. La distribución de intensidad máxima en ángulos grandes no es muy nítida debido a que las moléculas dentro de los planos esmécticos están dispuestas aleatoriamente. En el caso de las fases esmécticas C, el ángulo entre las capas esmécticas normales y el director (θ) ya no es colineal (Figura\(\PageIndex{40}\) d). Esta inclinación se puede ver fácilmente en el patrón de difracción, ya que los picos difusos en ángulos cada vez más pequeños ya no son ortogonales entre sí.
Preparación de Muestras
En general, las mediciones de dispersión de rayos X de muestras de cristal líquido se consideran más difíciles de realizar que las de muestras cristalinas. Se deben realizar los siguientes pasos para la medición por difracción de muestras de cristal líquido:
- La muestra debe estar libre de cualquier solvente y oxígeno absorbido, ya que su presencia afecta el carácter cristalino líquido de la muestra y su respuesta térmica. Esto se puede lograr realizando múltiples ciclos de fusión y congelación en vacío para eliminar disolventes y gases no deseados.
- Para realizar mediciones de baja resolución, la muestra de cristal líquido se puede colocar dentro de un capilar de vidrio de pared delgada. Los extremos del capilar pueden ser sellados por epoxi en caso de muestras volátiles. El proceso de llenado tiende a alinear las moléculas de cristal líquido a lo largo de la dirección del flujo.
- Para mediciones de alta resolución, la muestra generalmente está confinada entre dos cubreobjetos de vidrio recubiertos de polímero frotados separados por una junta tórica como espaciador. El frotamiento provoca la formación de surcos en la película de polímero que tiende a alinear las moléculas de cristal líquido.
- Las muestras alineadas son necesarias para identificar la fase cristalina líquida de la muestra. Las muestras de cristal líquido se pueden alinear calentando por encima de la temperatura de transición de fase y enfriándolas lentamente en presencia de un campo eléctrico o magnético externo. Un campo magnético es efectivo para muestras con núcleos aromáticos ya que tienen alta anisotropía diamagnética. Un problema común en el uso del campo eléctrico es el calentamiento interno que puede interferir con la medición.
- El tamaño de la muestra debe ser suficiente para evitar cualquier obstrucción al paso del haz de rayos X incidente.
- El grosor de la muestra debe estar alrededor de una longitud de absorción de los rayos X. Esto permite que alrededor del 63% de la luz incidente pase a través y obtenga una intensidad de dispersión óptima. Para la mayoría de los hidrocarburos, la longitud de absorción es de aproximadamente 1.5 mm con un objetivo metálico de cobre (λ = 1.5418 Å). El objetivo de molibdeno se puede utilizar para obtener una radiación de energía aún mayor (λ = 0.71069 Å).
Análisis de datos
La identificación de la fase de una muestra de cristal líquido es crítica para predecir sus propiedades físicas. Un simple patrón de difracción de rayos X 2D puede decir mucho a este respecto (Figura\(\PageIndex{40}\)). También es crítico determinar el orden orientacional de un cristal líquido. Esto es importante para caracterizar el grado de alineación de la muestra.
Por simplicidad, el resto de la discusión se centra en las fases de cristal líquido nemático. En una muestra no alineada, no hay ningún orden macroscópico específico en el sistema. En los dominios de tamaño micrométrico, todas las moléculas están orientadas en una dirección específica, llamada director local. Debido a que no hay orden posicional en los cristales líquidos nemáticos, este director local varía en el espacio y asume todas las orientaciones posibles. Por ejemplo, en una muestra perfectamente alineada de cristales líquidos nemáticos, todos los directores locales estarán orientados en la misma dirección. El alineamiento específico de moléculas en una dirección preferida en cristales líquidos hace que sus propiedades físicas tales como índice de refracción, viscosidad, susceptibilidad diamagnética, dependan direccionalmente.
Cuando una muestra de cristal líquido se orienta usando campos externos, los directores locales se alinean preferentemente globalmente a lo largo del director de campo. Esta dirección globalmente preferida se conoce como el director y se denota por el vector unitario n. La extensión de alineación dentro de una muestra de cristal líquido se denota típicamente por el parámetro de orden, S, como se define por\ ref {14}, donde θ es el ángulo entre el eje largo de la molécula y la dirección preferida, n.
\[ S\ =\ (\frac{3cos^{2} \theta \ -\ 1}{2}) \label{14} \]
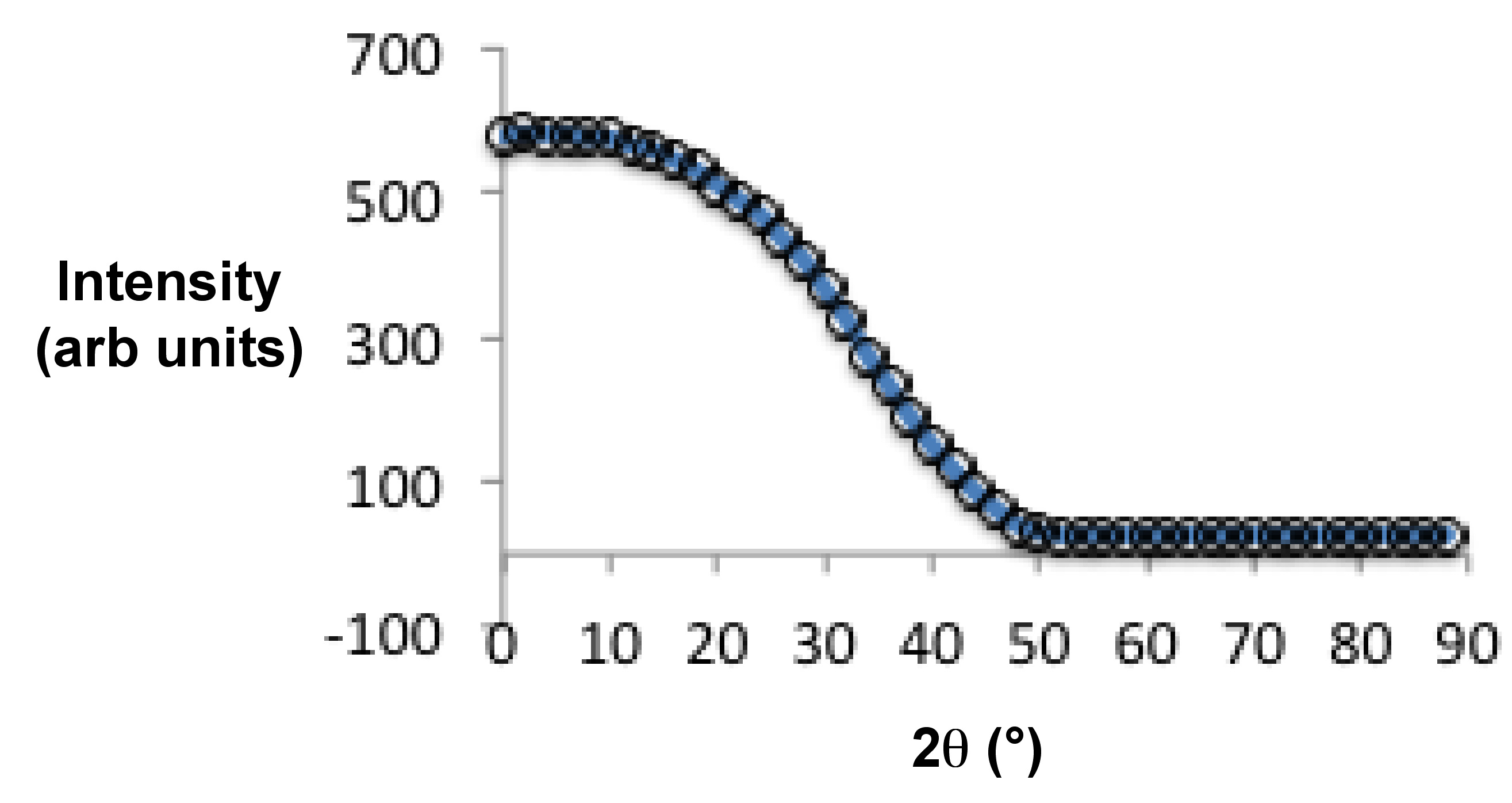
Para muestras isotrópicas, el valor de S es cero, y para muestras perfectamente alineadas es 1. La figura\(\PageIndex{41}\) muestra la estructura de una molécula de cristal líquido nemático más ampliamente estudiada, 4-ciano-4'-pentilbifenilo, comúnmente conocida como 5CB. Para la preparación de una muestra de polidominio se colocó 5CB dentro de un capilar de vidrio vía fuerzas capilares (Figura\(\PageIndex{41}\)). La Figura\(\PageIndex{42}\) muestra la difracción de rayos X 2D de la muestra de polidominio preparada. Para preparar la muestra de monodominio, se calentó un capilar de vidrio lleno de 5CB a 40 °C (es decir, por encima de la temperatura de transición nemático-isotrópica de 5CB, ~35 °C) y luego se enfrió lentamente en presencia de campo magnético (1 Testla, Figura\(\PageIndex{43}\). Esto da una muestra uniformemente alineada con el director nemático n orientado a lo largo del campo magnético. La figura\(\PageIndex{44}\) muestra la medición de difracción de rayos X 2D recolectada de una muestra de cristal líquido monodominio 5CB usando Rigaku Raxis-IV++, y consiste en dos arcos difusos (como se mencionó anteriormente). La figura\(\PageIndex{45}\) muestra la distribución de intensidad de un arco difuso en función de θ, y el valor calculado del parámetro de orden, S, es -0.48.




Refinamiento del Trastorno Cristalográfico en el Anión Tetrafluoroborato
A través de nuestra caracterización estructural de diversas sales de tetrafluoroborato, el catión complejo ha sido nominalmente el tema principal de interés; sin embargo, observamos que los aniones del anión tetrafluoroborato (BF 4 -) estaban comúnmente desordenados (13 de 23 estructuras investigado). Además, una consideración de la Base de Datos Estructurales de Cambridge al 14 de diciembre de 2010 arrojó 8,370 estructuras en las que está presente el anión tetrafluoroborato; de estas, 1044 (12.5%) fueron refinadas por tener algún tipo de trastorno asociado con el anión BF 4 -. Se han reportado varios métodos diferentes para el tratamiento de estos trastornos, pero la mayoría se refinó como una rotación no cristalográfica a lo largo del eje de uno de los enlaces B-F.
Desafortunadamente, la misma propiedad que hace que los fluoro-aniones sean tan buenos candidatos para contraiones no coordinantes (es decir, fuerzas intermoleculares débiles) también facilita la presencia de trastornos en las estructuras cristalinas. En otras palabras, la aparición del trastorno se intensifica con la presencia de un anión esférico débilmente coordinante (p. ej., BF 4 - o PF 6 -) que carece de las fuertes interacciones intermoleculares necesarias para mantener una orientación aniónica regular y repetitiva a lo largo del celosía cristalina. Esencialmente, estos aniones débilmente coordinados son esferas ricas en electrones débilmente definidas. Todos consideraron que parece que los fluoro-aniones, en general, tienen una propensión a exhibir parámetros aparentemente grandes de desplazamiento atómico (ADP's), y así, se refinan apropiadamente como teniendo ocupaciones fraccionarias de sitio.
Trastorno de refinación
En cristalografía, los parámetros de desplazamiento atómico observados son un promedio de millones de celdas unitarias en todo el volumen del cristal, y el movimiento inducido térmicamente a lo largo del tiempo utilizado para la recolección de datos. Un trastorno de átomos/moléculas en una estructura dada puede manifestarse como parámetros de desplazamiento atómico planos o no esféricos en la estructura cristalina. Tales casos de trastorno suelen ser el resultado de un movimiento inducido térmicamente durante la recolección de datos (es decir, trastorno dinámico), o del trastorno estático de los átomos/moléculas a lo largo de la red. Este último se define como la situación en la que ciertos átomos, o grupos de átomos, ocupan orientaciones ligeramente diferentes de molécula a molécula sobre el gran volumen (relativamente hablando) cubierto por la red cristalina. Este desplazamiento estático de átomos puede simular el efecto de la vibración térmica sobre el poder de dispersión del átomo “promedio”. En consecuencia, la diferenciación entre el movimiento térmico y el trastorno estático puede ser ambigua, a menos que la recolección de datos se realice a baja temperatura (lo que anularía gran parte del movimiento térmico observado a temperatura ambiente).
En la mayoría de los casos, este trastorno se resuelve fácilmente como algunos elementos de simetría no cristalográficos que actúan localmente sobre el anión débilmente coordinante. Las ocupaciones del sitio atómico se pueden refinar utilizando la instrucción FVAR en las diferentes partes (ver PARTE 1 y PARTE 2 en la Figura\(\PageIndex{47}\)) del trastorno, teniendo un factor de ocupación del sitio (s.o.f.) de x y 1-x, respectivamente. Esto se logra reemplazando 11.000 (en las líneas F-atom en el archivo “NAME.INS”) por 21.000 o -21.000 para cada una de las diferentes partes del trastorno. Por ejemplo, el archivo “NAME.INS” se vería algo así como el que se muestra en la Figura\(\PageIndex{47}\). Obsérvese que para estructuras más fuertemente desordenadas, es decir, aquellas con más de dos partes desordenadas, se puede utilizar el comando SUMP para determinar el s.o.f. de las partes 2, 3, 4, etc. cuya suma combinada se establece en s.o.f. = 1.0. Estos se designan en el FVAR como los términos segundo, tercero y cuarto.

En el refinamiento de moléculas pequeñas, inevitablemente surgirá el caso en el que se debe utilizar algún tipo de restricciones o restricciones para lograr la convergencia de los datos. Una restricción es cualquier información adicional relativa a una característica estructural dada, es decir, límites a los posibles valores de parámetros, que se puede agregar al refinamiento, aumentando así el número de parámetros refinados. Por ejemplo, los sistemas aromáticos son esencialmente planos, por lo que para fines de refinamiento, un sistema de anillo problemático podría restringirse para que se encuentre en un plano. Las restricciones no son exactas, es decir, están ligadas a una distribución de probabilidad, mientras que las restricciones son condiciones matemáticas exactas. Se puede considerar que las restricciones caen en uno de varios tipos generales:
- Restricciones geométricas, que relaciona distancias que deberían ser similares.
- Restricciones rígidas para grupos.
- Restricciones antichoques.
- Restricciones de parámetros vinculados.
- Restricciones de similitud.
- Restricciones ADP (Figura\(\PageIndex{48}\)
- Restricciones de suma y promedio.
- Fijación de origen y restricciones limitadoras de desplazamiento.
- Los impuestos sobre los parámetros de desplazamiento atómico.
Restricciones geométricas
- SADI - restricciones de distancia similares para pares de átomos nombrados.
- DFIX - restricción de distancia definida entre átomos unidos covalentemente.
- DANG - restricciones de distancia no enlazantes definidas, e.g., entre átomos F que pertenecen a la misma PARTE de un BF 4 desordenado -.
- PLANO - frena grupo de átomos para que se encuentren en un plano.
Limitaciones de parámetros de desplazamiento anisotrópico
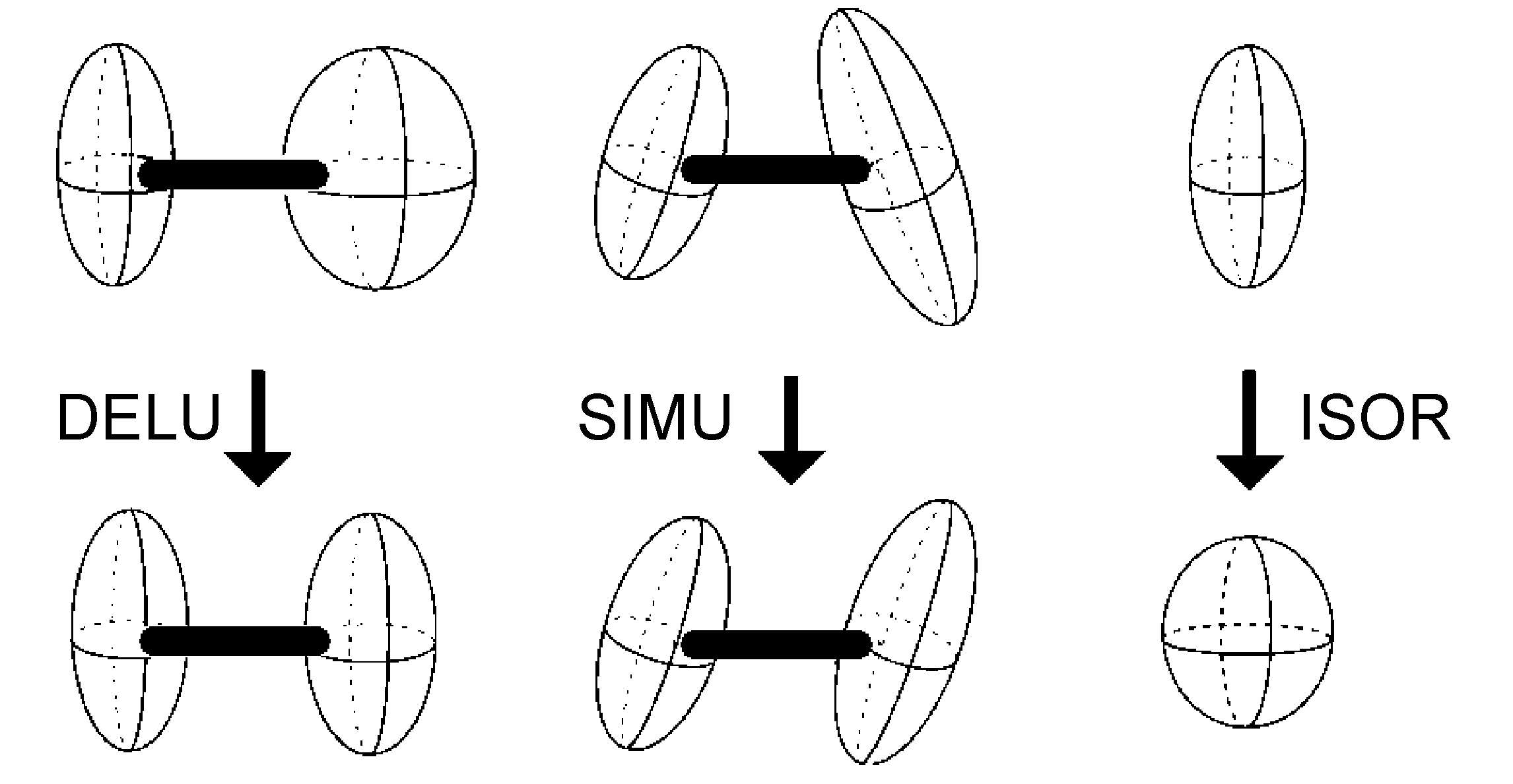
- DELU - restricciones de unión rígidas (Figura\(\PageIndex{48}\))
- SIMU - restricciones ADP similares en los componentes U ij correspondientes para ser aproximadamente iguales para átomos en estrecha proximidad (Figura\(\PageIndex{48}\))
- ISOR - tratar los átomos anisotrópicos nombrados para que tengan un comportamiento aproximadamente isotrópico (Figura\(\PageIndex{48}\))
Restricciones (distintas de las “restricciones”)
- EADP - parámetros de desplazamiento atómico equivalentes.
- AFIX - grupo ajustado; por ejemplo, AFIX 66 encajaría los siguientes seis átomos en un hexágono regular.
- HFIX - coloca los átomos de H en posiciones geométricamente ideales, por ejemplo, HFIX 123 colocaría dos conjuntos de átomos de H de metilo desordenados en dos sitios, a 180° entre sí.
Clase de Trastorno para el Anión Tetrafluoroborato
Rotación alrededor de un eje no cristalográfico a lo largo de un enlace B-F
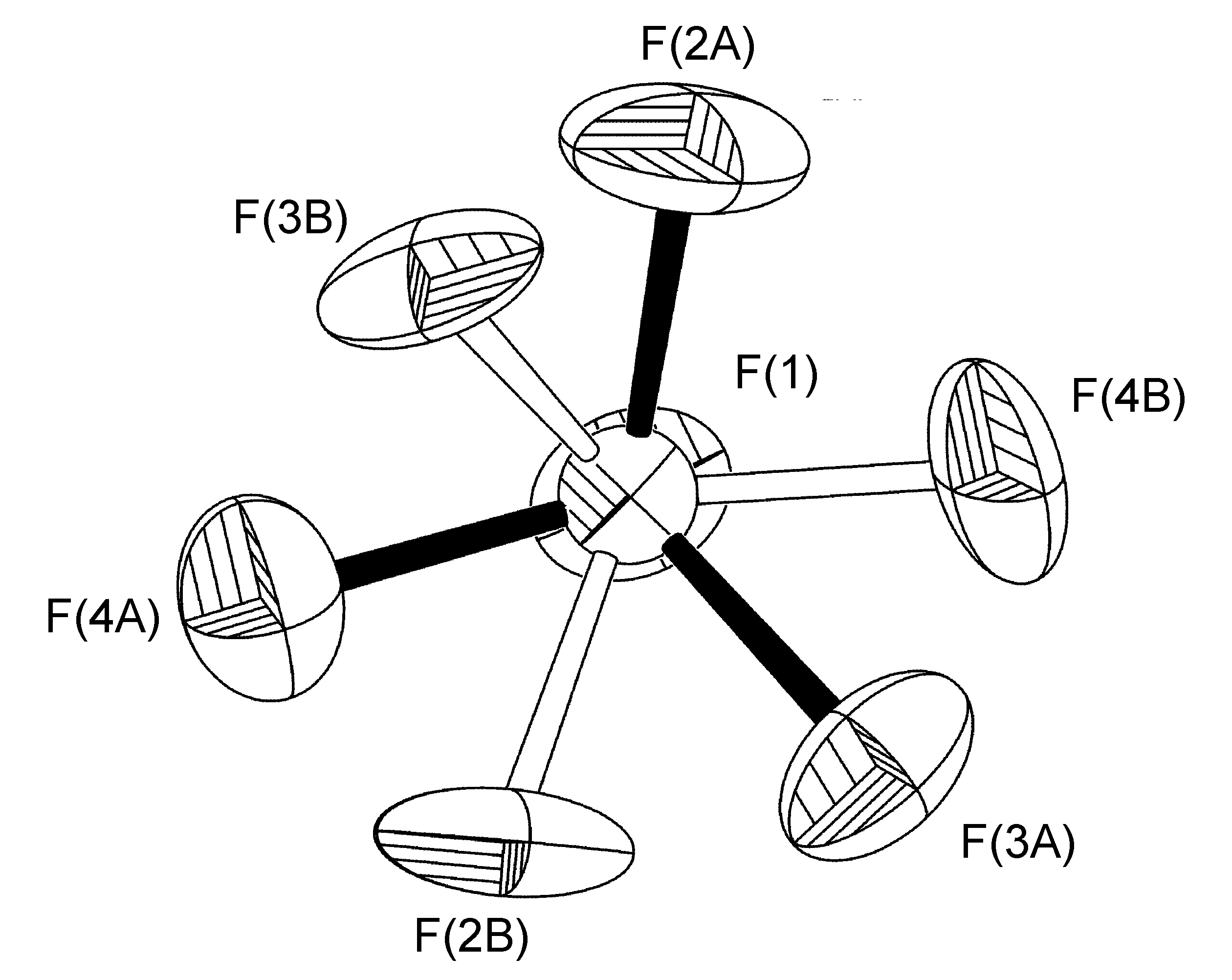
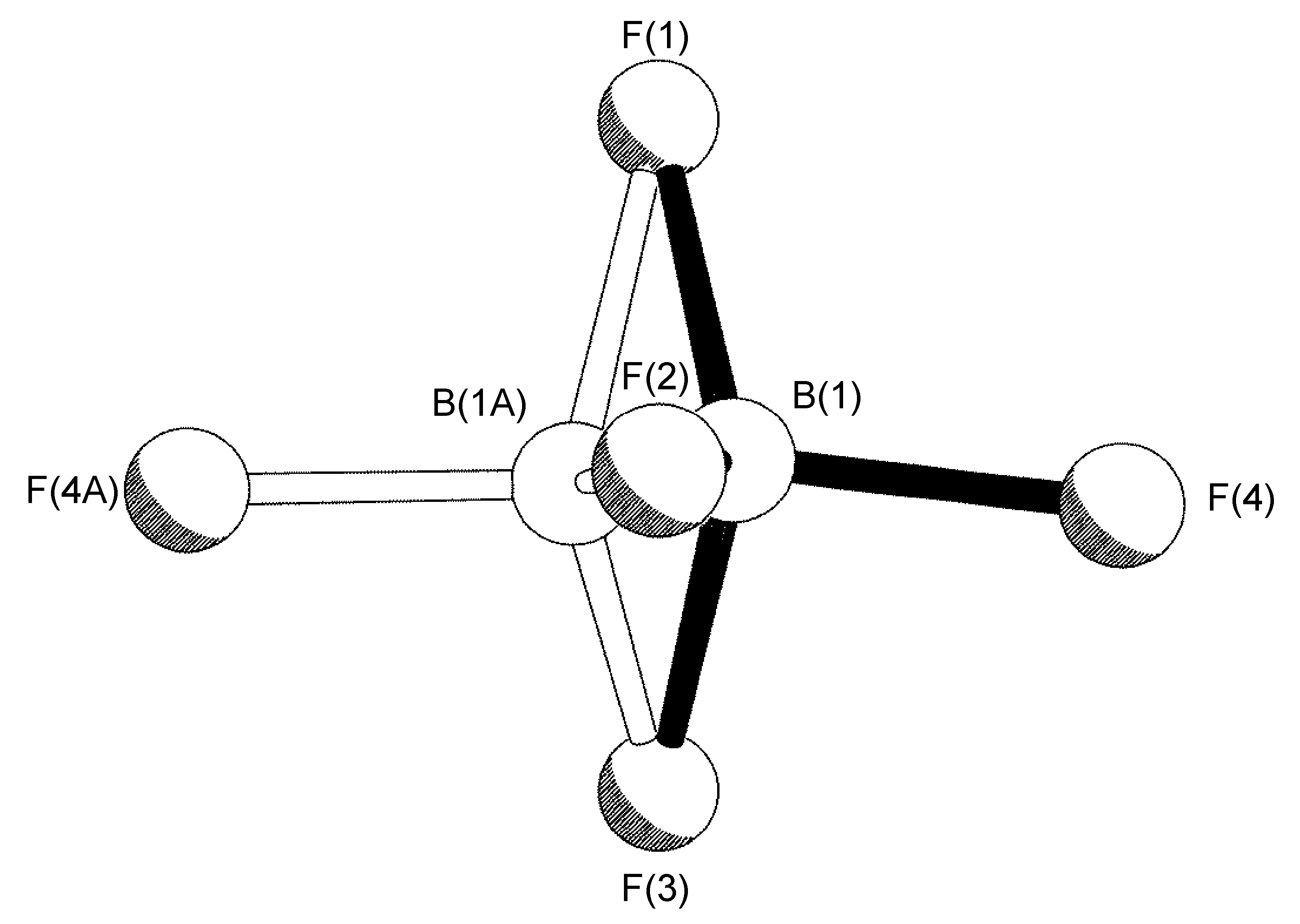
El caso más común de trastorno es una rotación alrededor de un eje, el más simple de los cuales involucra un eje de rotación relacionado con simetría no cristalográfica alrededor del vector hecho por uno de los enlaces B-F; esta operación conduce a que tres de los cuatro átomos F tengan dos ocupaciones de sitio (Figura\(\PageIndex{49}\)). Este trastorno también se observa para los grupos t Bu y CF 3, y debido a la simetría C 3 de los restos C (CH 3) 3, CF 3 y BF 3 realmente da como resultado una rotación cercana a C2.
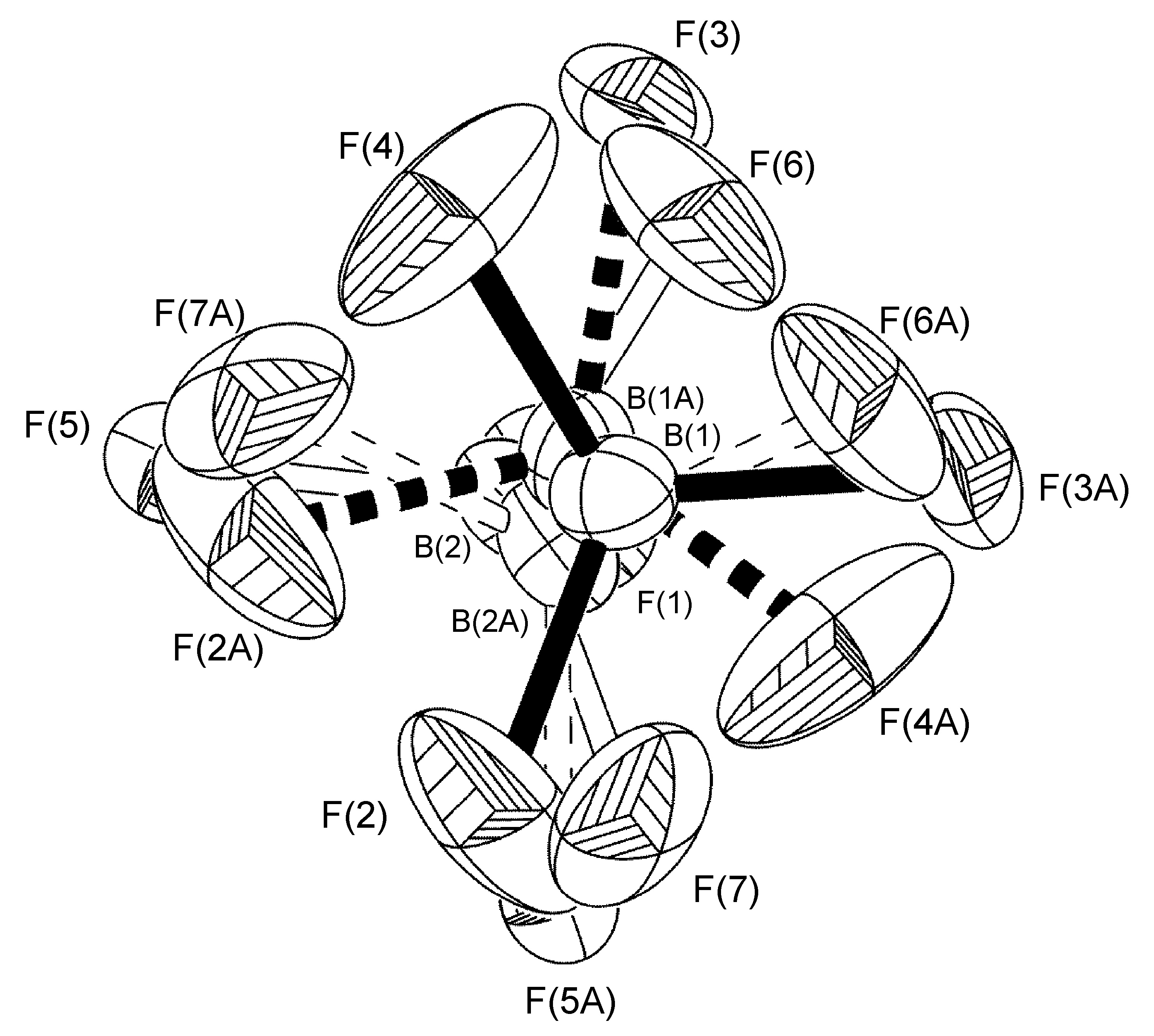
En un ejemplo típico, se encontró que el anión BF 4 - presente en la estructura cristalina de [H (MES-DPA)] BF 4 (Figura\(\PageIndex{50}\)) tenía un trastorno de ocupación del sitio 75:25 para tres de los cuatro átomos de flúor (Figura\(\PageIndex{51}\)). El trastorno es una rotación alrededor del eje del enlace B (1) -F (1). Para los ciclos iniciales de refinamiento, se colocaron restricciones de distancia similares (SADI) en todas las distancias B-F y F-F, además de restricciones ADP similares (SIMU) y restricciones de enlace rígido (DELU) para todos los átomos F. Se levantaron las restricciones para los ciclos finales de refinamiento. Se requirió un refinamiento de trastorno similar para [H (2- i PrpH-dPA)] BF 4 (45:55), mientras que el refinamiento del trastorno en [Cu (2- i PrPh-DPA) (estireno)] BF 4 (65:35) se realizó con solo las restricciones SADI y DELU se levantaron en ciclos finales de refinamiento.

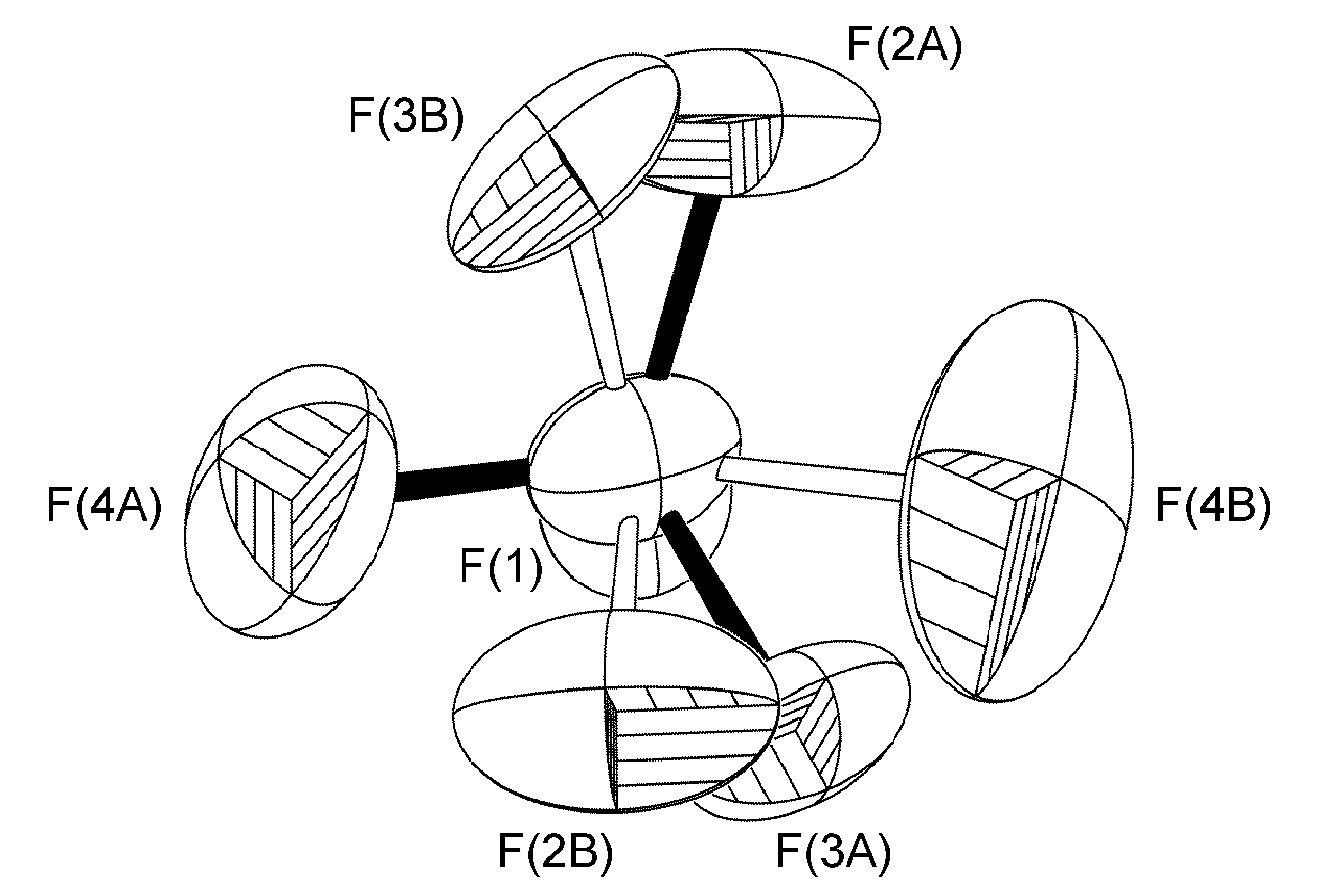
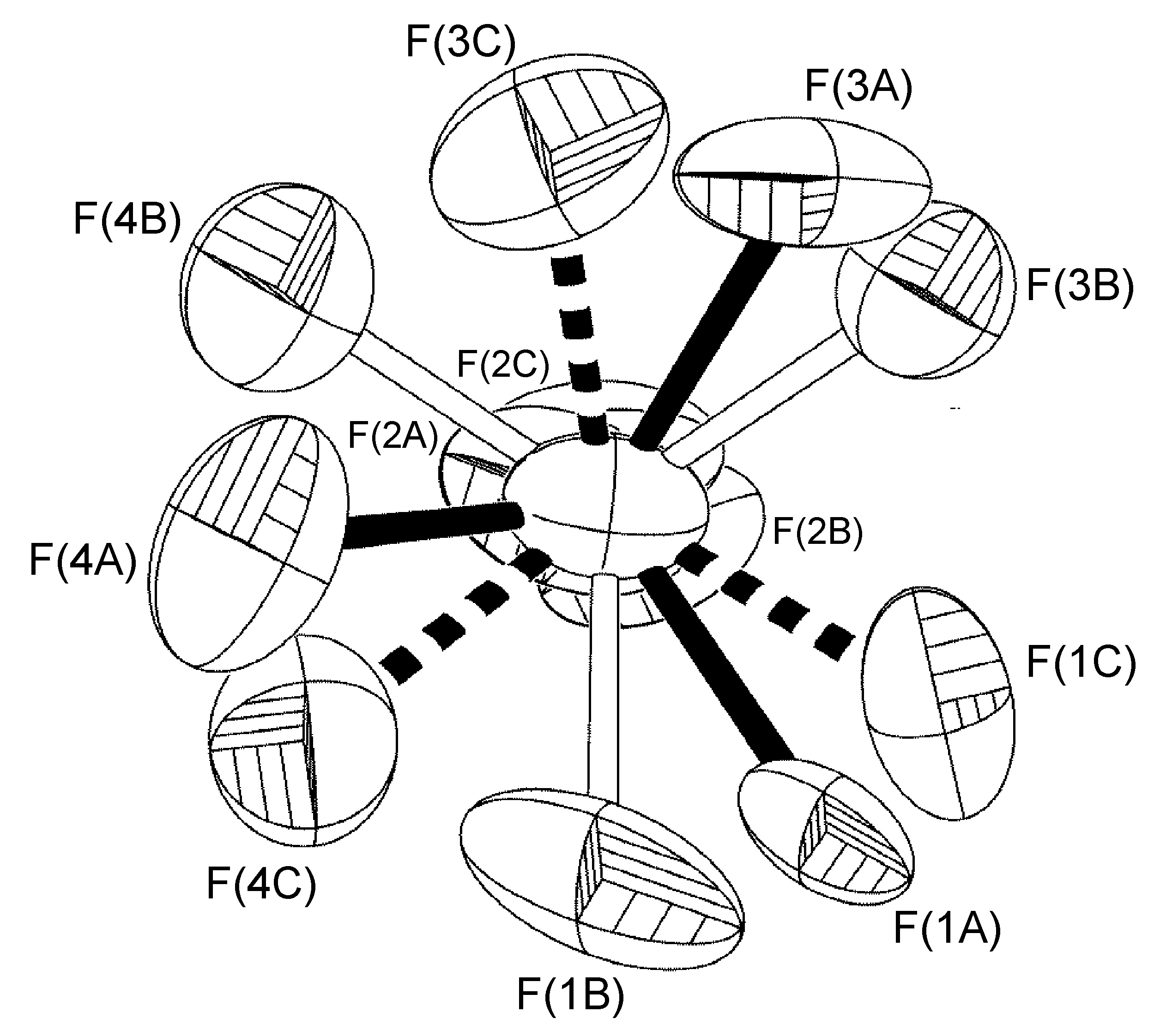
En el complejo [Ag (h-DPA) (estireno)] BF 4 el uso de la variable libre (FVAR) condujo al refinamiento de los átomos de flúor desordenados F (2A) -F (4A) y F (2B) -F (4B) por tener un trastorno de ocupación del sitio 75:25 (Figura\(\PageIndex{52}\)). Para los ciclos iniciales de refinamiento, todas las longitudes de enlace B-F recibieron restricciones de distancia similares (SADI). También se colocaron restricciones de distancia similares (SADI) en distancias F... F para cada parte, es decir, F (2A)... F (3A) = F (2B)... F (3B), etc. Adicionalmente, se colocaron restricciones ADP similares (SIMU) y restricciones de enlace rígido (DELU) en todos los átomos F. Todas las restricciones, con excepción del SIMU, fueron levantadas para los ciclos finales de refinamiento.
Rotación alrededor de un eje no cristalográfico, no a lo largo de un enlace B-F
El segundo tipo de trastorno está estrechamente relacionado con el primero, con la única diferencia de que el eje de rotación se inclina ligeramente del vector de enlace B-F, lo que resulta en que los cuatro átomos F tienen dos ocupaciones de sitio (Figura\(\PageIndex{53}\)). Los ángulos de inclinación varían de 6.5° a 42°.
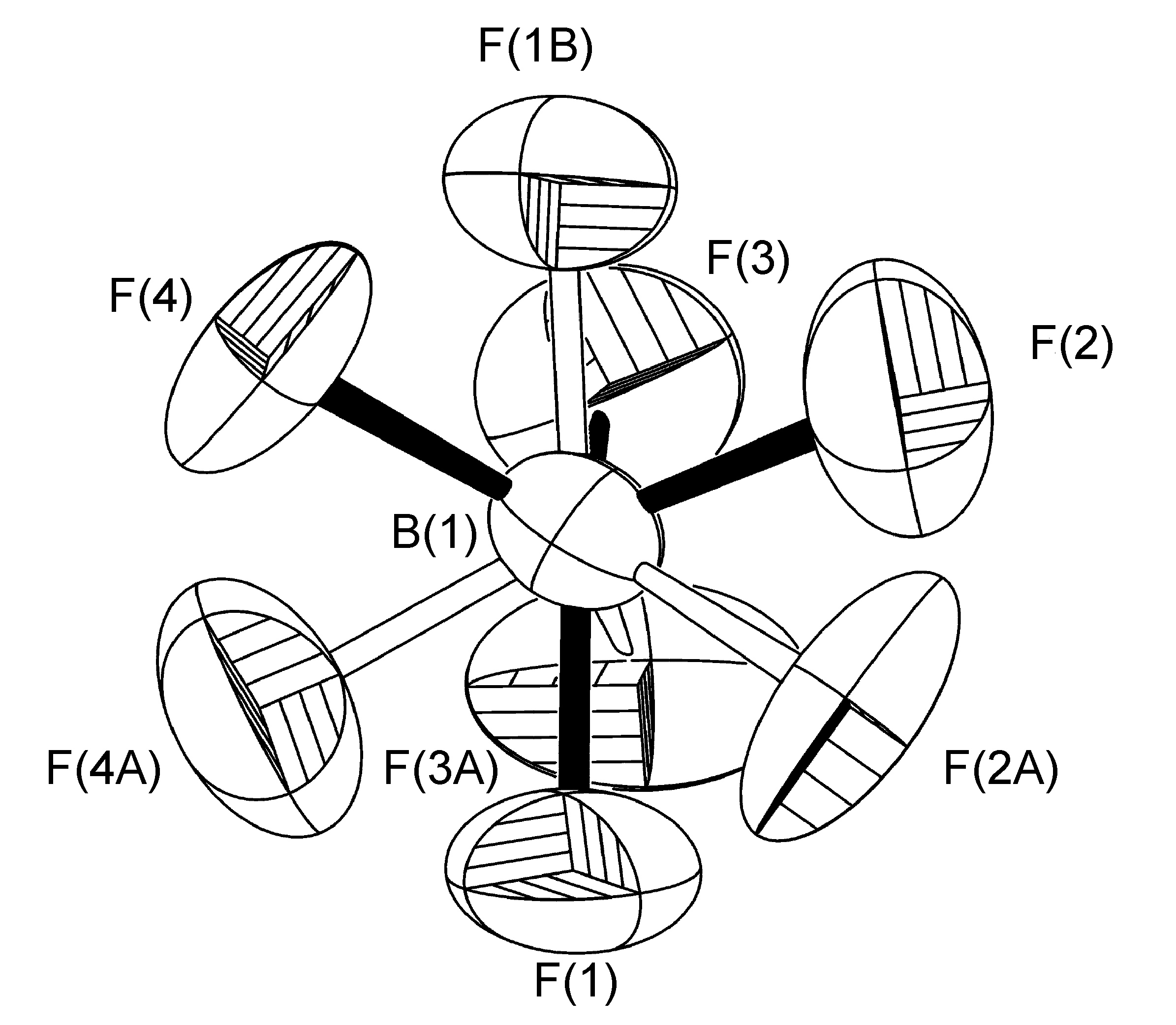
El anión BF 4 - desordenado presente en la estructura cristalina de [Cu (pH-dPA) (estireno)] BF 4 se refinó teniendo ocupaciones de sitio fraccional para los cuatro átomos de flúor alrededor de una rotación ligeramente inclinada del enlace B (1) -F (2A). Sin embargo, cabe señalar que si bien los valores de U (eq) determinados para los datos recopilados a baja temperatura son aproximadamente la mitad de los encontrados a temperatura ambiente, como es evidente por los tamaños y formas de los átomos de flúor en la Figura\(\PageIndex{54}\), las ocupaciones del sitio se refinaron a 50:50 en cada caso, y no hubo resolución en el desorden.
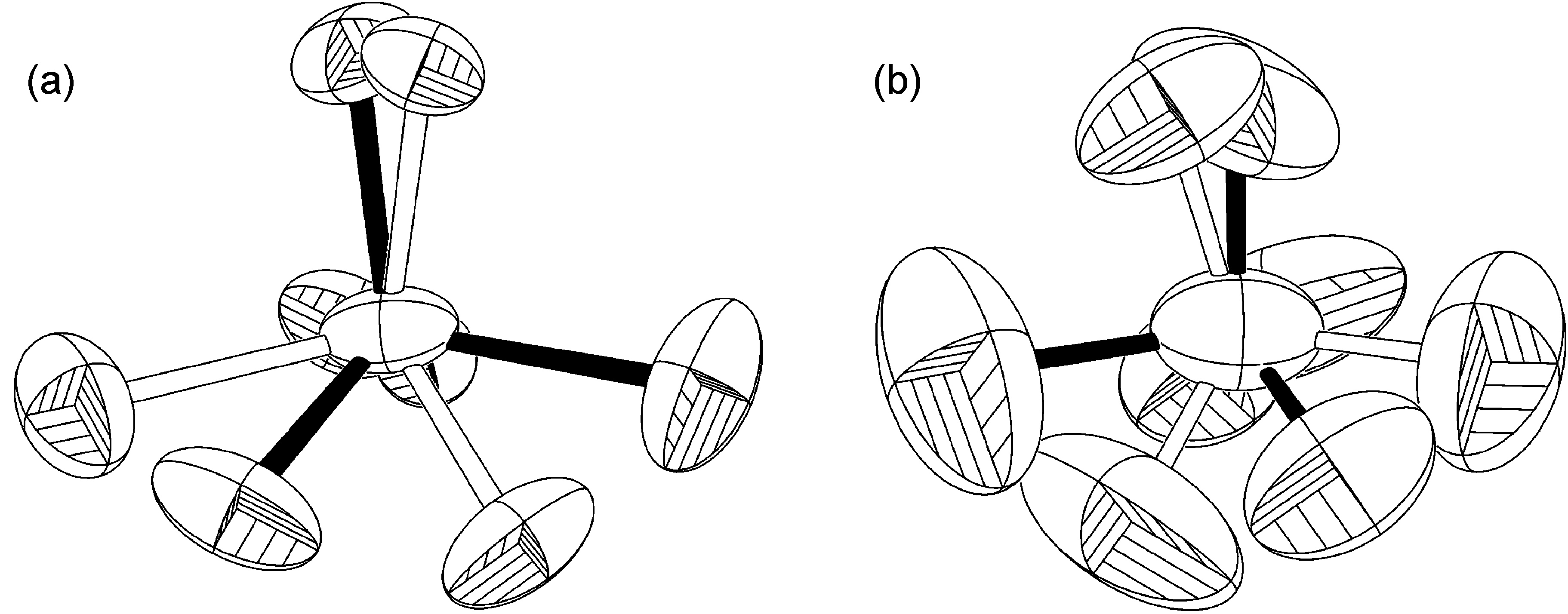
Se observa un ejemplo extremo de rotación fuera del eje donde el refinamiento de más de dos ocupaciones de sitio (Figura\(\PageIndex{55}\)) con hasta trece ubicaciones diferentes de átomos de flúor en un solo átomo de boro.
Rotación restringida alrededor de un eje no cristalográfico no a lo largo de un enlace B-F
Aunque es posible un amplio rango de ángulos de inclinación, en algunos sistemas el ángulo está limitado por la presencia de enlaces de hidrógeno. Por ejemplo, se encontró que el anión BF 4 - presente en [Cu (Mes-DPA) (μ-OH) (H 2 O)] 2 [BF 4] 2 tenía un trastorno de ocupación en el sitio 60:40 de los cuatro átomos de flúor, y mientras que el trastorno es una rotación C2 ligeramente inclinada hacia fuera del eje del enlace B (1) -F (1A), el ángulo está restringido por la presencia de dos interacciones B-F... O para uno de los isómeros (Figura\(\PageIndex{56}\)).
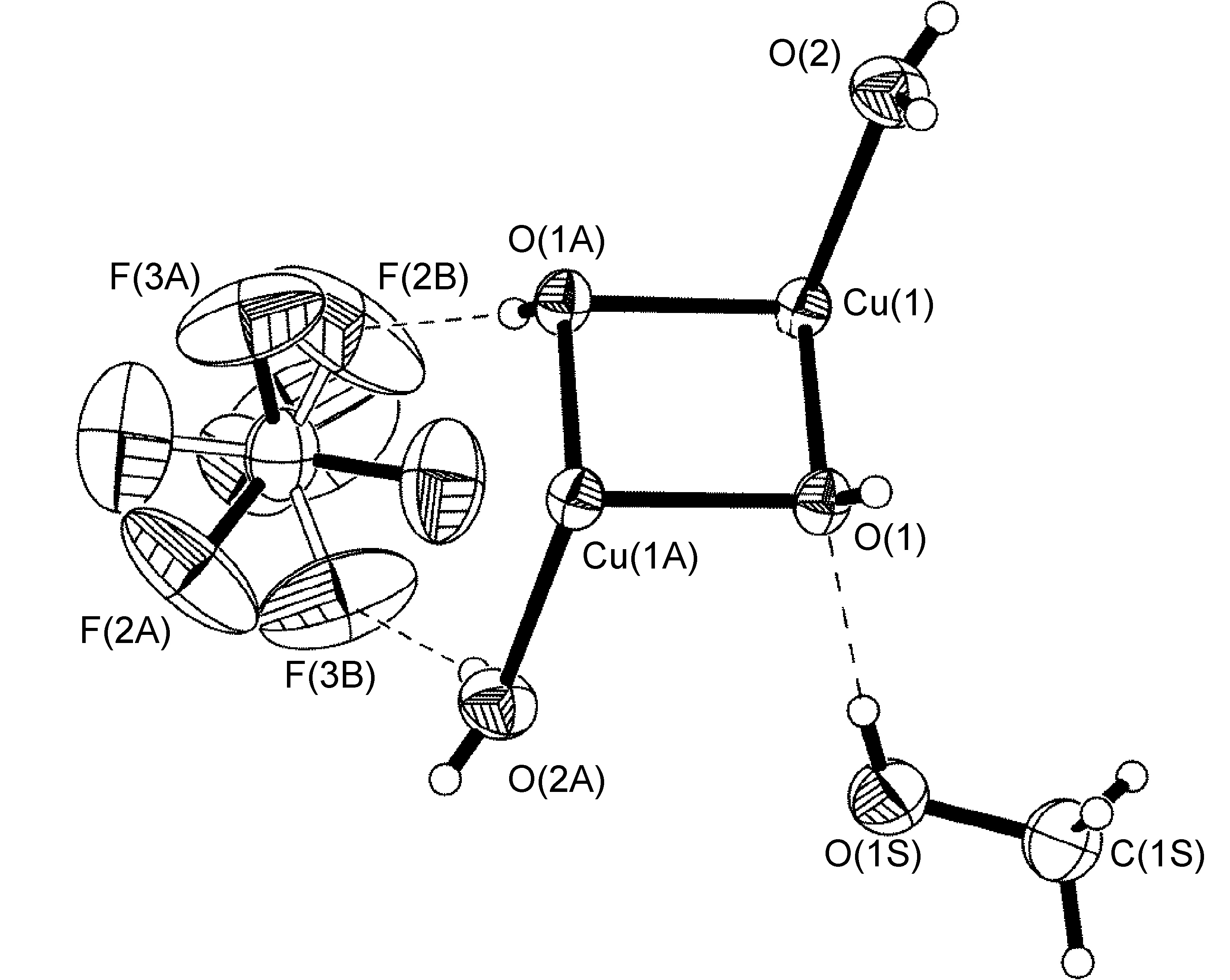
Un ejemplo que sí se adhiere a elementos de simetría global se ve en el anión BF 4 de [Cu {2,6- i Pr 2 C 6 H 3 N (quin) 2} 2] BF 4 .MeOH (Figura\(\PageIndex{57}\)), que exhibe un enlace de hidrógeno interacción con una molécula de disolvente de metanol desordenada. La estructura de R-N (quin) 2 se muestra en la Figura\(\PageIndex{54}\) b. Por simetría cristalográfica, el átomo de carbono del metanol y el átomo de boro del anión BF 4 se encuentran en un eje C2. Los átomos de flúor [F (1) -F (4)], el átomo de oxígeno del metanol y los átomos de hidrógeno unidos a los átomos de metanol O (1S) y C (1S) se refinaron teniendo un trastorno de ocupación del sitio 50:50 (Figura\(\PageIndex{57}\)).

Centro de Inversión No Cristalográfica en el Átomo de Boro
Se pueden observar múltiples trastornos con una célula unitaria monocristalina. Por ejemplo, los dos aniones BF 4 - en [Cu (Mes-DPA) (estireno)] BF 4 exhibieron trastornos de ocupación del sitio 50:50, el primero es una rotación C2 inclinada de uno de los enlaces B-F, mientras que el segundo está desordenado alrededor de una inversión centrada en el átomo de boro. El refinamiento de estos últimos se realizó de manera similar a los casos mencionados, con la excepción de que se dejaron en su lugar restricciones de distancia fija para los átomos no enlazados (DANG) para los átomos de flúor desordenados unidos a B (2) (Figura\(\PageIndex{58}\)).
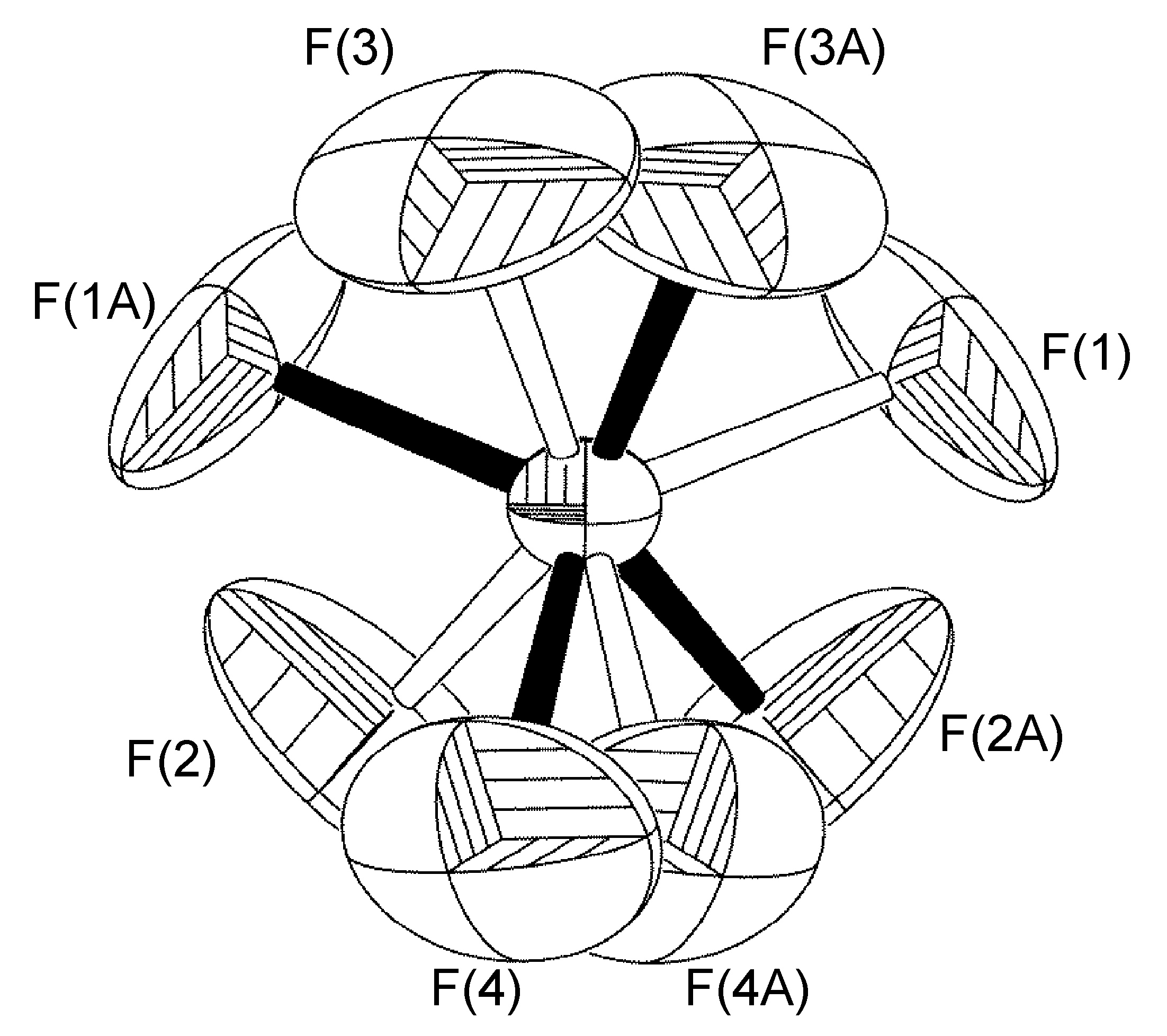
Trastorno en un plano especular cristalográfico
Otro caso en el que el anión BF 4 - está desordenado alrededor de un elemento de simetría cristalográfica es el de [Cu (h-dPA) (1,5-ciclooctadieno)] BF 4. En este caso los átomos de flúor F (1) a F (4) están presentes en la unidad asimétrica del complejo. Los átomos desordenados F (1A) -F (4A) se refinaron con 50% de ocupaciones de sitio, ya que B (1) se encuentra en un plano espejo (Figura\(\PageIndex{59}\)). Para los ciclos iniciales de refinamiento, se colocaron restricciones de distancia similares (SADI) en todas las distancias B-F y F-F, además de restricciones ADP similares (SIMU) y restricciones de enlace rígido (DELU) para todos los átomos F. Se levantaron las restricciones para los ciclos finales de refinamiento, en los que el átomo de boro se encuentra en un plano especular cristalográfico, y los cuatro átomos de flúor se reflejan a través de ellos.
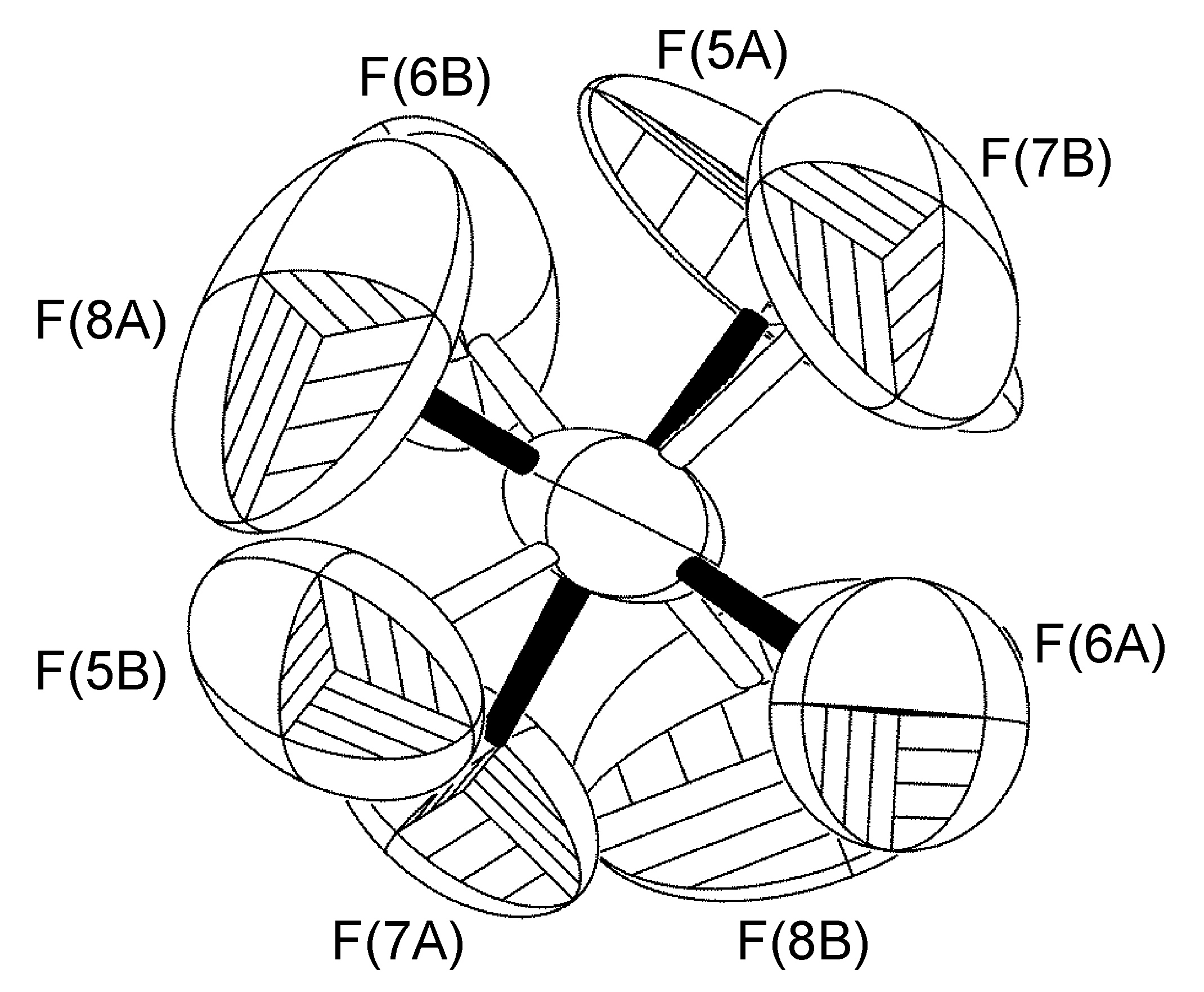
Trastorno en un plano especular no cristalográfico
Se ha observado que el anión BF 4 puede exhibir trastorno de ocupación del sitio del átomo de boro y uno de los átomos de flúor a través de un plano espejo NCS definido por el plano de los otros tres átomos de flúor (Figura\(\PageIndex{60}\)) modelando el anión completo como desordenado (incluyendo el átomo de boro).
Trastorno del núcleo del átomo de boro
El caso extremo de un trastorno implica el refinamiento de todo el anión, con todos los átomos de boro y todos los átomos de flúor ocupando más de dos sitios (Figura\(\PageIndex{61}\)). De hecho, algunos trastornos de estos últimos tipos deben ser refinados isotrópicamente, o como último recurso, en absoluto, para evitar que uno o más átomos se vuelvan definitivos no positivos.