7.8: Análisis de proteínas mediante espectroscopía de masas por ionización por electropulverización
- Page ID
- 70971

La ionización por electropulverización y espectrometría de masas (ESI-MS) es un método analítico que se enfoca en la determinación estructural macromolecular. El componente único de ESI-MS es la ionización por electronebulización. El desarrollo de la electropulverización, el proceso de carga de un líquido en un aerosol fino, se completó en la década de 1960 cuando Malcolm Dole (Figura\(\PageIndex{1}\)) demostró la capacidad de las especies químicas para separarse mediante técnicas de electropulverización. Con este importante giro de eventos, la combinación de ESI y EM fue factible y posteriormente fue desarrollada por John B. Fenn (Figura\(\PageIndex{2}\)), como un método analítico funcional que podría proporcionar información beneficiosa sobre la estructura y tamaño de una proteína. Fenn compartió el Premio Nobel en 2002, con Koichi Tanaka (Figura\(\PageIndex{3}\) y Kurt Wuthrich (Figura\(\PageIndex{4}\)) por el desarrollo de ESI-MS.




ESI-MS es el proceso a través del cual las proteínas, o macromoléculas, en la fase líquida se cargan y fragmentan en gotitas de aerosol más pequeñas. Estas gotitas de aerosol pierden su disolvente e impulsan los fragmentos cargados a la fase gaseosa en varios componentes que varían según la carga. Estos componentes pueden ser detectados por un espectrómetro de masas. El reciente auge y desarrollo de la ESI-MS se atribuye a sus beneficios en la caracterización y análisis de macromoléculas, específicamente macromoléculas biológicamente importantes como las proteínas.
¿Cómo funciona ESI-MS?
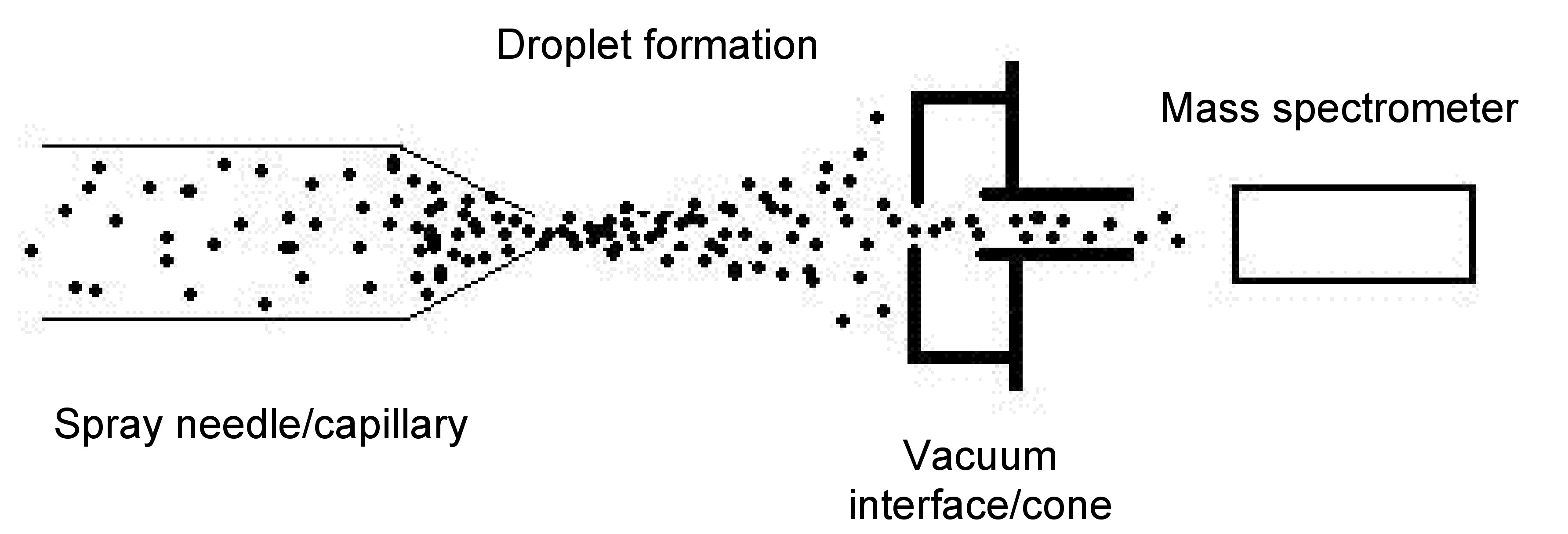
ESI-MS es un proceso que requiere que la muestra esté en solución líquida, de manera que pequeñas gotitas puedan ser ionizadas y analizadas individualmente por un espectrómetro de masas. A continuación se delinean los procesos que ocurren como relevantes para la Figura\(\PageIndex{5}\):
- Aguja de pulverización/capilar- La solución líquida de la macromolécula deseada se introduce en el sistema a través de esta aguja. La aguja está altamente cargada a través de una fuente de voltaje exterior que mantiene la carga constante a través de la aguja. La carga normal para una aguja es de aproximadamente 2.5 a 4 kV. El voltaje hace que las gotas grandes se fragmenten en pequeñas gotitas basadas en la carga que se acumula de las partes constituyentes de la proteína, y el líquido ahora está en fase gaseosa.
- Formación de gotitas- Las gotitas que son expulsadas de la aguja son más pequeñas que inicialmente, y como resultado el disolvente se evaporará. Luego, las gotitas más pequeñas comienzan a aumentar su densidad de carga en la superficie a medida que disminuye el volumen. Como las gotitas cerca del límite de Rayleigh, las interacciones coulómbicas de la gotita equivalen a la tensión superficial de la gotita, se produce una explosión coulómbica que rompe aún más la gotita en fracciones diminutas, incluyendo el analito aislado con carga.
- Interfaz/cono de vacío - Esta porción del dispositivo permite que las gotas se alineen en un pequeño rastro y pasen al espectrómetro de masas. La alineación ocurre debido a la similitud y diferencias en las cargas entre todas las gotitas. Todas las gotitas se ionizan a cargas positivas mediante la adición de protones a diferentes sitios básicos en las gotitas, sin embargo, todas las cargas varían en magnitud dependiendo del número de sitios básicos disponibles para la protonación. El extremo receptor o el cono tiene la carga opuesta de la aguja de pulverización, provocando una atracción entre el cono y las gotitas.
- Espectrómetro de masas- Las partículas cargadas alcanzan entonces el espectrómetro de masas y son desviadas en función de la carga de cada partícula. La deflexión ocurre por el imán cuadrupolar del espectrómetro de masas. Las diferentes trayectorias de deflexión de los iones ocurren debido a la fuerza de la interacción con el campo magnético. Esto conduce a varios caminos basados en una relación masa/carga (m/z). Las partículas son luego leídas por el detector de iones, a medida que llegan, proporcionando un espectro basado en la relación m/z.
¿Qué datos proporciona ESI-MS?
Como implica el nombre, los datos producidos a partir de esta técnica son un espectro de espectrometría de masas. Sin profundizar demasiado en el tema de la espectrometría de masas, que está fuera del verdadero alcance de este módulo, aquí se dará una ligera explicación. El espectrómetro de masas separa las partículas en base a un campo magnético creado por un imán cuadrupolar. La fuerza de la interacción varía según la carga que transportan las partículas. La cantidad de deflexión o fuerza de interacción es determinada por el detector de iones y cuantificada en una relación masa/carga (m/z). Debido a esta información, la determinación de la composición química o estructura peptídica puede manejarse fácilmente como se explica con mayor detalle en la siguiente sección.
Interpretación de un Espectro de EM Típico
Interpretar los datos de espectrometría de masas implica comprender la relación m/z. El conocimiento necesario para entender la interpretación del espectro es que los picos corresponden a porciones de toda la molécula. Es decir, hipotéticamente, si pones un cuerpo humano en el espectrómetro de masas, un pico coincidiría con un brazo, otro pico coincidiría con el brazo y el abdomen, etc. La idea general detrás de estos picos, es que una superposición pintaría todo el cuadro, o en el caso del hipotético ejemplo, proporcionar la imagen del cuerpo humano. La relación m/z define estas porciones en función de las cargas que llevan; de ahí la terminología de la relación masa/carga. Cuantas más cargas tenga una porción de la macromolécula o proteína, menor será la relación m/z y más a la izquierda aparecerá en el espectro. El concepto fundamental detrás de la interpretación implica comprender que los picos están interrelacionados, y así los cálculos matemáticos pueden realizarse para proporcionar información relevante de la proteína o macromolécula que se analiza.
Cálculos de m/z de los picos del espectro MS
Como se mencionó anteriormente, la información pertinente a obtener de los datos de ESI-MS es extrapolada a partir del entendimiento de que los picos están interrelacionados. Los pasos para calcular los datos son los siguientes:
- Determinar cuáles son los dos picos vecinos que se analizarán.
- Establecer el primer pico (el más lejano a la izquierda) como el pico con mayor relación m/z. Esto se define matemáticamente como nuestro pico z +1.
- Establecer el pico adyacente a la derecha de nuestro primer pico como el pico con la relación m/z más baja. Este es matemáticamente nuestro pico z.
- Nuestro pico z +1 también será nuestro pico m +1 ya que la diferencia entre los dos picos es la carga de un protón. En consecuencia, nuestro pico z se definirá como nuestro pico m.
- Resolver ambas ecuaciones para m para permitir la sustitución. Ambos lados de la ecuación deben estar en términos de z y pueden resolverse.
- Determinar la carga del pico z y posteriormente, la carga del pico z+1.
- Restar uno de la relación m/ z y multiplicar la relación m/z de cada pico por las cargas previas determinadas para obtener la masa de la proteína o macromolécula.
- Promedio de los resultados para determinar la masa promedio de la macromolécula o proteína.
1. Determinar qué dos picos vecinos se analizarán a partir de la MS (Figura\(\PageIndex{6}\)) como los picos m/z = 5 y m/z = 10.
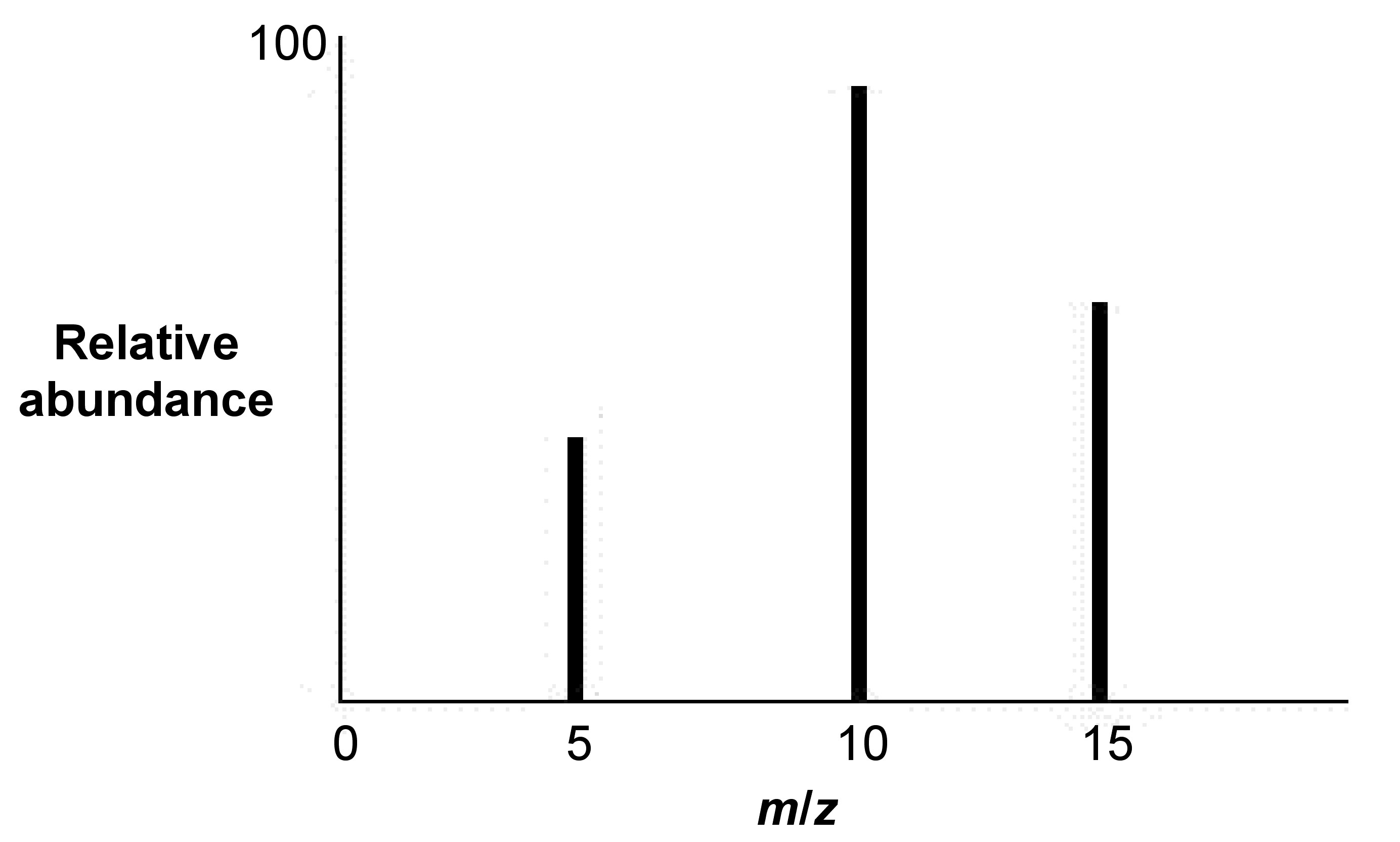
2. Establecer el primer pico (el más lejano a la izquierda en la Figura\(\PageIndex{1}\) como el pico z + 1 (es decir, z + 1 = 5).
3. Establecer el pico adyacente a la derecha del primer pico como el pico z (es decir, z = 10).
4. Establecer las relaciones de pico,\ ref {1} y\ ref {2}.
\[ \frac{m+1}{z+1} =\ 5 \label{1} \]
\[ \frac{m}{z} = 10 \label{2} \]
5. Resuelve las proporciones para m:\ ref {3} y\ ref {4}.
\[ m\ =\ 5z\ +\ 4 \label{3} \]
\[ m\ =\ 10z \label{4} \]
6. Sustituya una ecuación por m:\ ref {5}.
\[ 5z\ +\ 4\ =\ 10z \label{5} \]
7. Resuelve para z:\ ref {6}.
\[ z\ = 4/5 \label{6} \]
8. Encuentra z+1:\ ref {7}.
\[ z\ +\ 1\ =\ 9/5 \label{7} \]
Encuentra la masa molecular promedio restando la masa por 1 y multiplicando por la carga:\ ref {8} y\ ref {9}. De ahí que la masa promedio = 7.2
\[ (10\ -\ 1)(4/5)\ =\ 7.2 \label{8} \]
\[ (5\ -\ 1)(9/5)\ =\ 7.2 \label{9} \]
Preparación de Muestras
Las muestras para ESI-MS deben estar en estado líquido. Este requisito proporciona el medio necesario para cargar fácilmente las macromoléculas o proteínas en un estado de aerosol fino que puede fragmentarse fácilmente para proporcionar los resultados deseados. El beneficio de esta técnica es que las proteínas sólidas que antes fueron difíciles de analizar, como la metalotioneína, pueden disolverse en un disolvente apropiado que permitirá el análisis a través de ESI-MS. Debido a que la muestra se está suministrando al sistema como un líquido, el capilar puede cargar fácilmente la solución para comenzar la fragmentación de la proteína en fracciones más pequeñas La carga máxima del capilar es de aproximadamente 4 kV. Sin embargo, esta cantidad de carga no es necesaria para cada macromolécula. La carga apropiada depende del tamaño y las características del disolvente y de cada macromolécula individual. Esto ha permitido la eliminación del límite de peso molecular que alguna vez se mantuvo cierto para el análisis simple de espectrometría de masas de proteínas. Las proteínas grandes y macromoléculas ahora pueden detectarse y analizarse fácilmente a través de ESI-MS debido a la facilidad con la que las moléculas pueden fragmentarse.
Técnicas Relacionadas
Una técnica relacionada que se desarrolló aproximadamente al mismo tiempo que ESI-MS es la espectrometría de masas de desorción/ionización láser asistida por matriz (MALDI-MS). Esta técnica que se desarrolló a finales de la década de 1980 como pozos, tiene el mismo propósito fundamental; permitiendo el análisis de grandes macromoléculas mediante espectrometría de masas a través de una ruta alternativa de generación de la fase gaseosa necesaria para el análisis. En MALDI-MS, se usa una matriz, generalmente compuesta por ácido 3,5-dimetoxi-4-hidroxicinámico cristalizado (Figura\(\PageIndex{7}\)), agua y un disolvente organix, para mezclar el analito, y se usa un láser para cargar la matriz.

La matriz luego cocristaliza el analito y luego se utilizan pulsos del láser para provocar la desorción de la matriz y algunos de los cristales de analito con ella, lo que lleva a la ionización de los cristales y el cambio de fase al estado gaseoso. Los analitos son luego leídos por el espectrómetro de masas en tándem. La tabla compara\(\PageIndex{1}\) directamente algunos atributos entre ESI-MS y MALDI-MS. Cabe señalar que existen diversas variaciones tanto de ESI-MS como de MALDI-MS, variando los métodos de recolección de datos y el cuggy-backing de varios otros métodos (cromatografía líquida, electroforesis capilar, espectrometría de masas de plasma acoplado inductivamente, etc.), sin embargo, todos ellos tienen lo mismo principios fundamentales como estos dos métodos básicos.
| Detalles Experimentales | ESI-MS | MALDI-MS |
|---|---|---|
| Estado de analito inicial | Líquido | Líquido/sólido |
| Método de ionización | Aguja capilar cargada | Desorción láser matricial |
| Estado final del analito | Gas | Gas |
| Cantidad de proteína necesaria | 1 μL | 1 μL |
| Método de espectro | Espectrometría de masas | Espectrometría de masas |
Problemas con ESI-MS
ESI-MS ha demostrado ser útil en la determinación de la estructura terciaria y los cálculos de peso molecular de macromoléculas grandes. Sin embargo, todavía hay varios problemas incorporados con la técnica y el análisis de macromoléculas. Un problema es el aislamiento de la proteína deseada para su análisis. Si la proteína es incapaz de extraerse de la célula, esto se suele hacer a través de electroforesis en gel, existe un factor limitante en qué proteínas se pueden analizar. El citocromo c (Figura\(\PageIndex{7}\)) es un ejemplo de una proteína que puede aislarse y analizarse, pero proporciona una limitación interesante sobre cómo la técnica analítica no funciona para un análisis de proteínas completamente efectivo. El problema con el citocromo c es que aunque la proteína esté en su confirmación nativa, aún puede mostrar diferente distribución de carga. Esto ocurre debido a la disponibilidad de sitios básicos para la protonación que están constantemente expuestos al solvente. Cualquier ligero cambio en la conformación nativa puede provocar que se bloqueen sitios básicos, como en el citocromo c, causando que se vean diferentes proporciones m/z. Otra limitación interesante se observa cuando los elementos inorgánicos, como en las proteínas metalotioneínas que contienen zinc, se analizan mediante ESI-MS. Las metalotioneínas tienen varias isoformas que no muestran una tendencia consistente en los datos de ESI-MS entre las isoformas variadas. Las marcadas diferencias ocurren debido a que la metalación de cada isoforma es diferente, lo que provoca que la electropulverización y como resultado la protonación de la proteína sea diferente. Así, la incorporación de átomos metálicos en proteínas puede tener diversos efectos sobre los datos de ESI-MS debido a las interacciones inesperadas entre el centro metálico y la propia proteína.