
8.1: Caracterización de micropartículas mediante microscopía confocal
- Page ID
- 71298

Una breve historia de la microscopía confocal
La microscopía confocal fue inventada por Marvin Minsky (FIGURA) en 1957, y posteriormente patentada en 1961. Minsky estaba tratando de estudiar las redes neuronales para entender cómo aprenden los cerebros, y necesitaba una forma de imaginar estas conexiones en su estado natural (en tres dimensiones). Inventó el microscopio confocal en 1955, pero su utilidad no se realizó del todo hasta que la tecnología pudo ponerse al día. En 1973 Egger publicó las primeras células reconocibles, y los primeros microscopios comerciales se produjeron en 1987.

En la década de 1990, la microscopía confocal se volvió casi rutinaria debido a los avances en tecnología láser, fibra óptica, fotodetectores, recubrimientos dieléctricos de película delgada, procesadores de computadora, almacenamiento de datos, pantallas y fluoróforos. Hoy en día, la microscopía confocal es ampliamente utilizada en ciencias de la vida para estudiar células y tejidos.
Los fundamentos de la fluorescencia
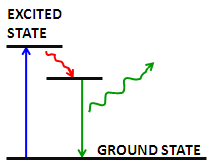
La fluorescencia es la emisión de un fotón secundario tras la absorción de un fotón de mayor longitud de onda. La mayoría de las moléculas a temperaturas normales se encuentran en el estado energético más bajo, el llamado 'estado de tierra'. Ocasionalmente, una molécula puede absorber un fotón y aumentar su energía al estado excitado. De aquí puede transferir muy rápidamente parte de esa energía a otras moléculas a través de colisiones; sin embargo, si no puede transferir suficiente energía emite espontáneamente un fotón con una figura de longitud de onda más baja\(\PageIndex{2}\). Esto es fluorescencia.
En la microscopía de fluorescencia, las moléculas fluorescentes están diseñadas para unirse a partes específicas de una muestra, identificándolas así cuando se obtienen imágenes. Se pueden utilizar múltiples fluoróforos para identificar simultáneamente diferentes partes de una muestra. Hay dos opciones al usar múltiples fluoróforos:
Se pueden elegir fluoróforos que respondan a diferentes longitudes de onda de un láser multilínea.
Se pueden elegir fluoróforos que respondan a la misma longitud de onda de excitación pero emitan a diferentes longitudes de onda.
Para aumentar la señal, se pueden unir más fluoróforos a una muestra. Sin embargo, hay un límite, ya que las altas concentraciones de fluoróforos dan como resultado que se apagen entre sí, y demasiados fluoróforos cerca de la superficie de la muestra pueden absorber suficiente luz para limitar la luz disponible para el resto de la muestra. Si bien se puede aumentar la intensidad de la radiación incidente, los fluoróforos pueden saturarse si la intensidad es demasiado alta.
El fotoblanqueo es otra consideración en la microscopía fluorescente. Los fluoróforos se desvanecen irreversiblemente cuando se exponen a la luz de excitación. Esto puede deberse a la reacción del estado excitado de las moléculas con oxígeno o radicales de oxígeno. Ha habido cierto éxito en la limitación del fotoblanqueo al reducir el oxígeno disponible o mediante el uso de depuradores de radicales libres. Algunos fluoróforos son más robustos que otros, por lo que la elección del fluoróforo es muy importante. Hoy en día están disponibles fluoróforos que emiten fotones con longitudes de onda que oscilan entre 400 y 750 nm.
En qué se diferencia la microscopía confocal de la microscopía óptica
Las lentes de un microscopio proyectan el plano de muestra sobre un plano de imagen. Se puede formar una imagen en muchos planos de imagen; sin embargo, solo consideramos que uno de estos planos es el 'plano focal' (cuando la imagen de muestra está enfocada). Cuando una pantalla estenopeica se coloca en el punto focal de la imagen, permite que pase la luz enfocada mientras bloquea efectivamente la luz de ubicaciones fuera de foco Figura\(\PageIndex{3}\). Este estenopeico se coloca en el plano de imagen conjugada al plano focal, de ahí el nombre “confocal”. El tamaño de este estenopeico determina la profundidad de enfoque; un orificio más grande recoge la luz de un volumen mayor. El estenopeico solo puede hacerse prácticamente tan pequeño como aproximadamente el radio del disco Airy, que es el mejor punto de luz posible a partir de una abertura circular Figura\(\PageIndex{4}\), porque más allá de esa más señal se bloquea dando como resultado una disminución de la relación señal/boise.
En óptica, el disco Airy y el patrón Airy son descripciones del punto de luz mejor enfocado que puede hacer una lente perfecta con una apertura circular, limitado por la difracción de la luz.
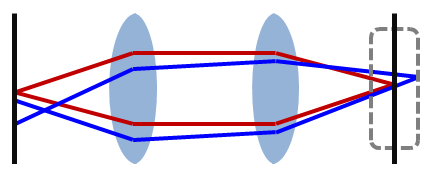

Para reducir aún más el efecto de dispersión debido a la luz de otras partes de la muestra, la muestra solo se ilumina en un punto minúsculo mediante el uso de un agujero de alfiler frente a la fuente de luz. Esto reduce en gran medida la interferencia de la luz dispersada de otras partes de la muestra. La combinación de un agujero de alfiler frente tanto a la fuente de luz como al detector es lo que hace que el confocal sea único.
Partes de un Microscopio Confocal
Un microscopio confocal simple generalmente consiste en un láser, una abertura estenopeica, un espejo dicromático, espejos de escaneo, objetivos de microscopio, un tubo fotomultiplador y un software de computación utilizado para reconstruir la imagen Figura\(\PageIndex{5}\). Debido a que un volumen relativamente pequeño de la muestra se está iluminando en un momento dado, se debe usar una fuente de luz muy brillante para producir una señal detectable. Los primeros microscopios confocales utilizaron lámparas de arco de circonio, pero los recientes avances en la tecnología láser han hecho que los láseres en los rayos UV-Visible e infrarrojos sean más estables y Un láser permite una fuente de luz monocromática (rango de longitud de onda estrecha) que se puede utilizar para excitar selectivamente fluoróforos para emitir fotones de una longitud de onda diferente. A veces, los filtros se utilizan para detectar más longitudes de onda individuales.
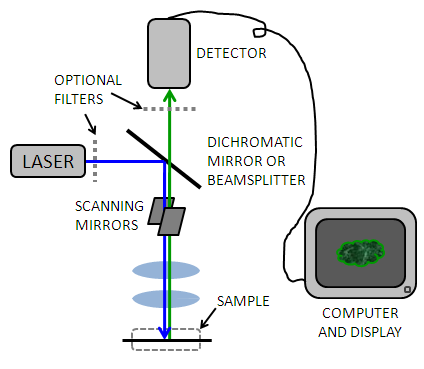
La luz pasa a través de una figura de espejo dicromático (o “dicroico”)\(\PageIndex{6}\) que permite pasar luz con una longitud de onda mayor (desde el láser) pero refleja luz de menor longitud de onda (de la muestra) al detector. Esto permite que la luz recorra la misma trayectoria a través de la mayor parte del instrumento, y elimina la señal debido a la reflexión de la luz incidente.
Luego, la luz se refleja a través de un par de espejos o cristales, uno cada uno para las direcciones x e y, que permiten que el haz escanee a través de la muestra (Figura\(\PageIndex{6}\)). La velocidad del escaneo suele ser el factor limitante en la velocidad de adquisición de imágenes. La mayoría de los microscopios confocales pueden crear una imagen en 0.1 - 1 segundo. Por lo general, la muestra se escanea ráster rápidamente en la dirección x y lentamente en la dirección y (como leer un párrafo de izquierda a derecha, Figura\(\PageIndex{6}\)).
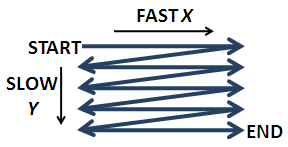
El rastering es controlado por galvanómetros que mueven los espejos hacia adelante y hacia atrás en un movimiento de diente de sierra. La desventaja de escanear con el haz de luz es que el ángulo de luz que golpea la muestra cambia. Afortunadamente, este cambio es pequeño. Curiosamente, el diseño original de Minsky movió el escenario en lugar del haz, ya que era difícil mantener la alineación de la óptica sensible. A pesar de las obvias desventajas de mover un espécimen voluminoso, hay algunas ventajas de mover el escenario y mantener la óptica estacionaria:
La luz ilumina el espécimen axialmente en todas partes evitando aberraciones ópticas, y
El campo de visión se puede hacer mucho más grande controlando la amplitud de los movimientos del escenario.
Una alternativa a los espejos reflectantes de luz es el deflector acústico-óptico (AOD). El AOD permite exploraciones rápidas en la dirección x mediante la creación de una rejilla de difracción a partir de ondas sonoras estacionarias de alta frecuencia (presión) que cambian localmente el índice de refracción de un cristal. La desventaja de los AOD es que la cantidad de deflexión depende de la longitud de onda, por lo que la luz de emisión no puede ser desnatada (viajar de regreso por la misma trayectoria que la luz de excitación). La solución a esto es desescanear solo en la dirección y controlada por el galvanómetro lento y recoger la luz en una hendidura en lugar de en una estenopeica. Esto da como resultado un corte óptico reducido y una ligera distorsión debido a la pérdida de simetría radial, pero aún se pueden formar buenas imágenes. ¡Tenga en cuenta que esto no es un problema para la microscopía de luz reflejada que tiene la misma longitud de onda para la luz incidente y reflejada!
Otra alternativa es el disco Nipkow, que tiene una matriz espiral de picaduras que crean el muestreo simultáneo de muchos puntos en la muestra. Una sola rotación cubre todo el espécimen varias veces (a 40 revoluciones por segundo, es decir, más de 600 cuadros por segundo). Esto permite el descanso, pero solo alrededor del 1% de la luz de excitación pasa a través. Esto está bien para la microscopía de luz reflejada, pero la señal es relativamente débil y la relación señal/ruido es baja. Los orificios podrían hacerse más grandes para aumentar la transmisión de la luz, pero luego la sección óptica es menos efectiva (recuerde que la profundidad de campo depende del diámetro del orificio) y la resolución xy es más pobre. Con este método se necesitan fluoróforos eficientes y altamente receptivos.
Volviendo al microscopio confocal (Figura\(\PageIndex{5}\)), la luz pasa luego por el objetivo que actúa como una combinación de condensador y objetivo bien corregida. Los fluoróforos iluminados emiten fluorescencia y la luz emitida viaja por el objetivo de regreso al espejo dicromático. Esto se conoce como epifluorescencia cuando la luz incidente tiene la misma trayectoria que la luz detectada. Dado que la luz emitida ahora tiene una longitud de onda menor que la incidente, no puede pasar a través del espejo dicromático y es reflejada hacia el detector. Cuando se usa luz reflejada, se usa un divisor de haz en lugar de un espejo dicromático. La microscopía de fluorescencia cuando se usa correctamente puede ser más sensible que la microscopía de luz reflejada.
Aunque la posición de la señal está bien definida de acuerdo con la posición de los espejos xy, la señal de fluorescencia es relativamente débil después de pasar a través del orificio, por lo que se utiliza un tubo fotomultiplador para detectar los fotones emitidos. Detectar todos los fotones sin tener en cuenta la posición espacial aumenta la señal, y el tubo fotomultiplador aumenta aún más la señal de detección al propagar una cascada de electrones resultante del efecto fotoeléctrico (fotones incidentes que arrancan electrones). La señal resultante es una señal eléctrica analógica con voltaje continuamente variable que corresponde a la intensidad de emisión. Esto es muestreado periódicamente por un convertidor analógico-digital.
Es importante entender que la imagen es una reconstrucción de muchos puntos muestreados a través del espécimen. En un momento dado el microscopio sólo está mirando un punto minúsculo, y no existe ninguna imagen completa que pueda verse en un punto instantáneo en el tiempo. El software se utiliza para recombinar estos puntos para formar un plano de imagen y combinar planos de imagen para formar una representación tridimensional del volumen de muestra.
Microscopía de dos fotones
La microscopía de dos fotones es una técnica mediante la cual dos haces de menor intensidad se dirigen para cruzarse en el punto focal. Dos fotones pueden excitar un fluoróforo si lo golpean al mismo tiempo, pero por sí solos no tienen suficiente energía para excitar ninguna molécula. La probabilidad de que dos fotones golpeen un fluoróforo casi exactamente al mismo tiempo (menos de 10-16) es muy baja, pero más probable en el punto focal. Esto crea un punto de luz brillante en la muestra sin el cono de luz habitual por encima y por debajo del plano focal, ya que casi no hay excitaciones alejadas del punto focal.
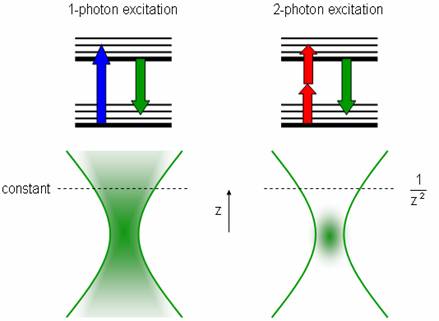
Para aumentar las posibilidades de absorción, se utiliza un láser pulsado ultrarrápido para crear pulsos de luz rápidos e intensos. Dado que la forma del reloj de arena es reemplazada por una fuente puntual, se puede eliminar el orificio cerca del detector (utilizado para reducir la señal de la luz que se origina desde fuera del plano focal). Esto también aumenta la relación señal/ruido (aquí hay muy poco ruido ahora que la fuente de luz está tan enfocada, pero la señal también es pequeña). Estos láseres tienen una potencia de incidente promedio menor que los láseres normales, lo que ayuda a reducir el daño a la muestra circundante. Esta técnica puede obtener imágenes más profundas en el espécimen (~400 μm), pero estos láseres siguen siendo muy caros, difíciles de configurar, requieren una fuente de alimentación más fuerte, enfriamiento intensivo y deben alinearse en la misma tabla óptica porque los pulsos pueden distorsionarse en las fibras ópticas.
Caracterización de micropartículas
La microscopía confocal es muy útil para determinar las posiciones relativas de las partículas en tres dimensiones Figura\(\PageIndex{8}\). El software permite la medición de distancias en las reconstrucciones 3D para que se pueda determinar la información sobre el espaciado (como densidad de empaque, porosidad, orden o alineación de largo alcance, etc.).
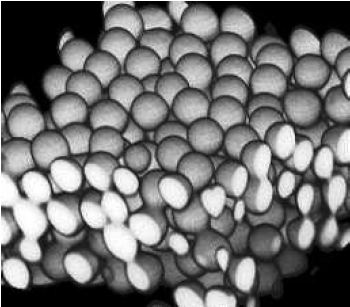
Figura\(\PageIndex{8}\) Una reconstrucción de una suspensión coloidal de micropartículas de poli (metacrilato de metilo) (PMMA) de aproximadamente 2 micras de diámetro. Adaptado de Microscopía Confocal de Coloides, Eric Weeks.
Si toma imágenes en modo de fluorescencia, recuerde que la señal solo representará las ubicaciones de los fluoróforos individuales. No hay garantía de que los fluoróforos se adhieran completamente a las estructuras de interés o que no habrá fluoróforos desviados lejos de esas estructuras. Para las micropartículas a menudo es posible unir los fluoróforos a la cubierta de la partícula, creando esferas huecas de fluoróforos. Es posible saber si una esfera de muestra es hueca o sólida pero dependería de la transparencia del material.
Las dispersiones de micropartículas se han utilizado para estudiar la nucleación y el crecimiento de cristales, ya que los coloides son mucho más grandes que los átomos y se pueden obtener imágenes en tiempo real. Las regiones cristalinas se determinan a partir del orden de las esferas dispuestas en una red, y las regiones se pueden distinguir entre sí al anotar defectos de la red.
El autoensamblaje es otra aplicación donde los estudios 3-D dependientes del tiempo pueden ayudar a dilucidar el proceso de ensamblaje y determinar la posición de varias estructuras o materiales. Debido a que el confocal es popular para especímenes biológicos, se puede observar la posición de nanopartículas como puntos cuánticos en una célula o tejido. Esto puede ser útil para determinar toxicidad, efectividad de administración de fármacos, limitaciones de difusión, etc.
Un resumen de las fortalezas y debilidades de la microscopía confocal
Fortalezas
Menos neblina, mejor contraste que los microscopios ordinarios.
Capacidad 3-D.
Ilumina un pequeño volumen.
Excluye la mayor parte de la luz de la muestra no en el plano focal.
La profundidad de campo se puede ajustar con el tamaño del estenopeico.
Tiene modos tanto de luz reflejada como de fluorescencia.
Puede obtener imágenes de células y tejidos vivos.
La microscopía de fluorescencia puede identificar varias estructuras diferentes simultáneamente.
Acomoda muestras con espesores de hasta 100 μm.
Se puede utilizar con microscopía de dos fotones.
Permite el seccionamiento óptico (sin artefactos de la sección física) 0.5 - 1.5 μm.
Debilidades
Las imágenes se escanean lentamente (una imagen completa cada 0.1-1 segundo).
Debe escanear la muestra ráster, no existe ninguna imagen completa en un momento dado.
Existe un límite de resolución inherente debido a la difracción (basada en la apertura numérica, ~200 nm).
La muestra debe ser relativamente transparente para una buena señal.
Las altas concentraciones de fluorescencia pueden apagar la señal fluorescente.
Fluoróforos fotoblanqueadores irreversiblemente.
Los láseres son caros.
El ángulo de la luz incidente cambia ligeramente, introduciendo una ligera distorsión.