
3.5: Aplicaciones - Cinética de Reacciones Catalíticas
- Page ID
- 69196

Es posible predecir cómo la cinética de ciertas reacciones catalizadas heterogéneamente podría variar con las presiones parciales de los gases reactivos por encima de la superficie del catalizador utilizando la expresión de isoterma de Langmuir para coberturas superficiales de equilibrio.
Descomposición Unimolecular
Considere la descomposición superficial de una molécula A, es decir, el proceso
\[A_{(g)} \rightleftharpoons A_{(ads)} → Products\]
Supongamos que:
- La reacción de descomposición se produce uniformemente a través de los sitios superficiales en los que la molécula A puede adsorberse y no se limita a un número limitado de sitios especiales.
- Los productos están muy débilmente ligados a la superficie y, una vez formados, se desorben rápidamente.
- El paso de determinación de la velocidad (rds) es el paso de descomposición de la superficie.
En estas circunstancias, las moléculas de A adsorbidas en la superficie están en equilibrio con las de la fase gaseosa y podemos predecir la concentración superficial de A a partir de la isoterma de Langmuir, i.e.
\[θ = \dfrac{bP}{1 + bP}\]
La velocidad de descomposición de la superficie (y por lo tanto de la reacción) viene dada por una expresión de la forma
\[rate = k θ\]
Esto supone que la descomposición de A (anuncios) ocurre en una etapa de reacción elemental unimolecular simple y que la cinética es de primer orden con respecto a la concentración superficial de este intermedio adsorbido). Sustituyendo la cobertura, θ, nos da la expresión requerida para la tasa en términos de la presión del gas sobre la superficie
\[rate = \dfrac{k b P}{1 + b P} \label{rate}\]
Es útil considerar dos extremos:
Baja presión/límite de unión
Este es el límite de baja presión (o unión débil. es decir, pequeño\(b\)): bajo estas condiciones la cobertura superficial en estado estacionario\(θ\),, de la molécula reaccionante es muy pequeña.
\[b P \ll 1\]
entonces
\[1 + bP \approx 1\]
y la ecuación se\(\ref{rate}\) puede simplificar a
\[rate \approx kbP\]
Bajo este caso limitante, la cinética sigue una reacción de primer orden (con respecto a la presión parcial de\(A\)) con una constante de velocidad aparente de primer orden\(k' = kb\).
Alta presión/límite de unión
Este es el límite de alta presión (o unión fuerte, es decir, grande\(b\)): bajo estas condiciones la cobertura superficial en estado estacionario,\(θ\), de la molécula reaccionante es casi la unidad y
\[bP \gg 1\]
entonces
\[1 + bP \approx bP\]
y la ecuación se\(\ref{rate}\) puede simplificar a
\[rate \approx k\]
bajo este caso limitante, la cinética sigue una reacción de orden cero (con respecto a la presión parcial de\(A\)). La tasa muestra la misma variación de presión que la cobertura superficial, pero esto no es sorprendente ya que es directamente proporcional a θ.
Estos dos casos limitantes pueden identificarse en la cinética general a partir de la Ecuación\(\ref{rate}\) en la Figura 3.5.1.
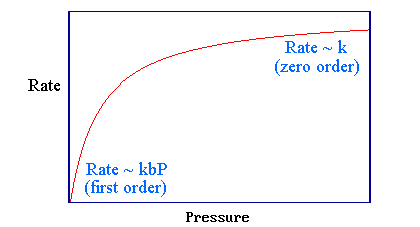
Figura 3.5.1:
Reacción bimolecular (entre adsorbatos moleculares)
Considere una reacción de Langmuir-Hinshelwood del siguiente tipo:
\[A_{(g)} \rightleftharpoons A_{(ads)} \label{Eq2.1}\]
\[B_{(g)} \rightleftharpoons B_{(ads)} \label{Eq2.2} \]
\[A_{(ads)} + B_{(ads)} \overset{slow}{\longrightarrow} AB_{(ads)} \overset{fast}{\longrightarrow} AB_{(g)} \label{Eq2.3} \]
Asumiremos además, como se señala en el esquema anterior, que la reacción superficial entre las dos especies adsorbidas (lado izquierdo de la Ecuación\(\ref{Eq2.3}\) es el paso determinante de la velocidad.
Si las dos moléculas adsorbidas son móviles sobre la superficie y se entremezclan libremente, entonces la velocidad de la reacción vendrá dada por la siguiente expresión de velocidad para la etapa de combinación de superficie bimolecular
\[Rate = k θ_A θ_B\]
Para un solo adsorbato molecular, la cobertura superficial (dada por la isoterma estándar de Langmuir) es
\[ θ = \dfrac{bP}{1 + bP}\]
Cuando dos moléculas (\(A\)&\(B\)) compiten por los mismos sitios de adsorción, entonces las expresiones relevantes son (ver derivación):
\[\theta_A = \dfrac{b_AP_A}{1+b_AP_A + b_BP_B}\]
y
\[\theta_B = \dfrac{b_BP_B}{1+b_AP_A + b_BP_B}\]
Sustituyendo estos en la expresión de la tasa da:
\[Rate = k \theta_A \theta_B = \dfrac{k b_AP_A b_BP_B }{( 1+b_AP_A + b_BP_B )^2}\]
Una vez más, es interesante mirar varios límites extremos
Baja presión/límite de unión
\[b_A P_A \ll 1\]
y
\[b_B P_B \ll 1\]
En este límite que\(θ_A\) &\(θ_B\) son ambos muy bajos, y
\[rate → k b_A P_A b_B P_B = k' P_A P_B\]
es decir, primer orden en ambos reactivos
Presión mixta/límite de unión
\[b_A P_A \ll 1 \ll b_B P_B\]
En este límite\(θ_A → 0\),\(θ_B → 1\), y
\[Rate → \dfrac{k b_A P_A }{b_B P_B } = \dfrac{k' P_A}{P_B}\]
es decir, primer orden en\(A\), pero primer orden negativo en\(B\)
Claramente, dependiendo de la presión parcial y la fuerza de unión de los reactivos, un modelo dado para el esquema de reacción puede dar lugar a una variedad de cinéticas aparentes: esto pone de relieve los peligros inherentes al proceso inverso, es decir, tratar de usar datos cinéticos para obtener información sobre la reacción mecanismo.
En superficies de metales preciosos (por ejemplo, Pt), generalmente se cree que la reacción de\(CO\) oxidación se realiza mediante un mecanismo Langmuir-Hinshelwood del siguiente tipo:
\[CO_{(g)} \rightleftharpoons CO_{(ads)}\]
\[O_{2 (g)} \rightleftharpoons 2 O_{(ads)}\]
\[CO_{(ads)} + O_{(ads)} \overset{slow}{\longrightarrow} CO_{2 (ads)} \overset{fast}{\longrightarrow} CO_{2 (g)}\]
Como el CO 2 está relativamente débilmente unido a la superficie, la desorción de esta molécula producto es relativamente rápida y en muchas circunstancias es la reacción superficial entre las dos especies adsorbidas la que es la etapa determinante de la velocidad.
Si se supone que las dos moléculas adsorbidas son móviles sobre la superficie y se entremezclan libremente, entonces la velocidad de la reacción vendrá dada por la siguiente expresión de velocidad para la etapa de combinación de superficie bimolecular
\[Rate = k \,θ_{CO}\, θ_O\]
Cuando dos especies de este tipo (una de las cuales está adsorbida molecularmente y la otra adsorbida disociativamente) compiten por los mismos sitios de adsorción, entonces las expresiones relevantes son (ver derivación):
\[\theta_{CO} = \dfrac{b_{CO}P_{CO}}{1+ \sqrt{b_OP_{O_2}} + b_{CO}P_{CO}}\]
y
\[\theta_{O} = \dfrac{ \sqrt{b_OP_{O_2}} }{1+ \sqrt{b_OP_{O_2}} + b_{CO}P_{CO}}\]
Sustituyendo estos en la expresión de la tasa da:
\[rate = k \theta_{CO} \theta_O = \dfrac{ k b_{CO}P_{CO} \sqrt{b_OP_{O_2}} }{(1+ \sqrt{b_OP_{O_2}} + b_{CO}P_{CO})^2} \label{Ex1.1}\]
Una vez más, es interesante mirar ciertos límites. Si el\(CO\) está mucho más fuertemente ligado a la superficie de tal manera que
\[b_{CO}P_{CO} \gg 1 + \sqrt{b_OP_{O_2}}\]
entonces
\[1 + \sqrt{b_OP_{O_2}} + b_{CO}P_{CO} \approx b_{CO}P_{CO}\]
y la Ecuación\(\ref{Ex1.1}\) simplifica para dar
\[rate \approx \dfrac{k \sqrt{b_OP_{O_2}} } {b_{CO}P_{CO}} = k' \dfrac{P^{1/2}_{O_2}}{P_{CO}}\]
En este límite las cinéticas son de orden medio con respecto a la presión en fase gaseosa del oxígeno molecular, pero de orden negativo con respecto a la presión\(CO\) parcial, es decir,\(CO\) actúa como veneno (a pesar de ser un reactivo) y aumentar su presión ralentiza la reacción. Esto se debe a que el CO está tan fuertemente unido a la superficie que bloquea la adsorción de oxígeno, y sin suficientes átomos de oxígeno en la superficie se reduce la velocidad de reacción.