
13.1: Movimientos rotacionales de Moléculas Rígidas
- Page ID
- 74372

En el Capítulo 3 y el Apéndice G se describen los niveles de energía y las funciones de onda que describen la rotación de moléculas rígidas. Por lo tanto, en este Capítulo estos resultados se resumirán brevemente y se pondrá énfasis en detallar cómo se obtienen las ecuaciones rotacionales correspondientes de Schrödinger y los supuestos y limitaciones subyacentes a las mismas.
Moléculas Lineales
Como se indica en el Capítulo 3, la ecuación de Schrödinger para el movimiento angular de una molécula diatómica rígida (es decir, que tiene una longitud de enlace fija R) es
\[ \dfrac{\hbar^2}{2\mu}\left[ \dfrac{1}{R^2 \sin \theta}\dfrac{\partial}{\partial \theta} \left( \sin \theta \dfrac{\partial}{\partial \theta} \right) + \left( \dfrac{1}{R^2\sin^2\theta} \right) \dfrac{\partial^2}{\partial \phi^2} \right] \psi = E \psi \]
o más sucintamente en términos del operador de momento angular como
\[ \dfrac{L^2\psi}{2\mu R^2} = E \psi \]
El hamiltoniano en este problema contiene solo la energía cinética de rotación; no hay energía potencial presente porque la molécula está experimentando una “rotación libre” sin obstáculos. Los ángulos\( \theta \text{ and } \phi\) describen la orientación del eje de la molécula diatómica en relación con un sistema de coordenadas fijado en laboratorio, y\(\mu\) es la masa reducida de la molécula diatómica
\[\mu=\dfrac{m_1m_2}{m_1+m_2}.\]
Las funciones propias y los valores propios
Los valores propios correspondientes a cada función propia son sencillos de encontrar porque\(H_{rot}\) son proporcionales al\(L^2\) operador cuyos valores propios ya han sido determinados. Las energías rotacionales resultantes se dan como:
\[ E_J = \hbar ^2\dfrac{J(J+1)}{(2\mu R^2)} = BJ(J+1) \]
y son independientes de\(M\). Así, cada nivel de energía está etiquetado por\(J\) y es\(2J+1\) -fold degenerado (porque\(M\) va desde\(-J\) hasta\(J\)). La constante rotacional B (definida como\(\hbar^2/2\mu R^2\) depende de la longitud del enlace de la molécula y la masa reducida. Las separaciones entre los niveles rotacionales sucesivos (que son de relevancia espectroscópica porque las reglas de selección de momento angular\(\Delta J\) a menudo se restringen a 1,0 y -1) vienen dadas por
\[ \Delta E = B(J+1)(J+2) - BJ(J+1) = 2B(J+1). \]
Dentro de este modelo de “rotor rígido”, el espectro de absorción de una molécula diatómica rígida debe mostrar una serie de picos, cada uno de los cuales corresponde a una\(J \rightarrow J + 1\) transición específica. Las energías a las que ocurren estos picos deben crecer linealmente con J. Un ejemplo de tal progresión de líneas rotacionales se muestra en la figura siguiente.
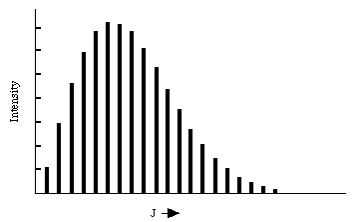
Las energías a las que ocurren las transiciones rotacionales parecen ajustarse bastante bien a la\(\Delta E = 2B (J+1)\) fórmula. Las intensidades de las transiciones del nivel J al nivel J+1 varían fuertemente con J principalmente debido a que la población de moléculas en el nivel absorbente varía con J. Estas poblaciones PJ se dan, cuando el sistema está en equilibrio a temperatura T, en términos de la degeneración (2J+1) del\(J^{th}\) nivel y la energía de este nivel B J (J+1):
\[ P_J = Q^{-1}(2J+1) e^{\dfrac{-BJ(J+1)}{kT}}, \]
donde\(Q\) está la función de partición rotacional:
\[ Q = \sum\limits_J (2J+1)e^{\dfrac{-BJ(J+1)}{kT}}. \]
Para valores bajos de\(J\), la degeneración es baja y el\(e^{\frac{-BJ(J+1)}{kT}}\) factor está cerca de la unidad. A medida que aumenta J, la degeneración crece linealmente pero el\(e^{\frac{-BJ(J+1)}{kT}}\) factor disminuye más rápidamente. En consecuencia, existe un valor de J, dado tomando la derivada de\((2J+1)e^{- \frac{BJ(J+1)}{kT}}\) respecto a J y poniéndola igual a cero,
\[ 2J_max + 1 = \sqrt{\dfrac{2kT}{B}} \]
en el que se espera que la intensidad de la transición rotacional alcance su máximo.
Las funciones propias que pertenecen a estos niveles de energía son los armónicos esféricos\(Y_{L,M}(\theta ,\phi)\) que se normalizan de acuerdo con
\[ \int\limits^\pi_0 \int\limits^{2\pi}_0 Y^{\text{*}}_{L,M}(\theta ,\phi) Y_{L',M'}(\theta ,\phi) sin\theta d\theta d\phi = \delta_{L,L'}\delta_{M,M'}. \]
Estas funciones son idénticas a las que aparecen en la solución de la parte angular de los átomos similares a hidrógeno. Los niveles de energía y funciones propias anteriores también se aplican a la rotación de moléculas poliatómicas lineales rígidas; la única diferencia es que el momento de inercia que\(I\) entra en la expresión de energía rotacional viene dado por
\[ I = \sum \limits_am_a R_a^2 \]
donde\(m_a\) esta la masa del\(a^{th} \text{ atom and } R_a\) es su distancia del centro de masa de la molecula. Este momento de inercia reemplaza\(mR^2\) en las expresiones de nivel de energía rotacional anteriores.
Moléculas no lineales
El operador de energía cinética rotacional para una molécula poliatómica rígida se muestra en el Apéndice G como
\[ H_{rot} = \dfrac{J_b^2}{2I_a} + \dfrac{J_b^2}{2I_b} + \dfrac{J_c^2}{2I_c}\]
donde los\(I_k\) (k = a, b, c) son los tres momentos principales de inercia de la molécula (los valores propios del tensor momento de inercia). Este tensor tiene elementos en un sistema de coordenadas cartesianas (K, K' = X, Y, Z) cuyo origen se localiza en el centro de masa de la molécula que se computan como:
\[ I_{K,K} \sum\limits_j m_j (R_j^2 - R^2_{K,j}) (\text{for K = K'}) \]
\[ I_{K,K'} = -\sum\limits_jm_jR_{K,j}R_{K',j} (\text{ for K }\neq\text{ K'}). \]
Los componentes de los operadores de momento angular mecánico cuántico a lo largo de los tres ejes principales son:
\[ J_a = -i\hbar \cos \chi \left[ \cot \theta \dfrac{\partial}{\partial \chi} - \dfrac{1}{\sin \theta} \dfrac{\partial}{\partial \phi} \right] - -i\hbar \sin\chi \dfrac{\partial}{\partial \theta} \]
\[ J_b = i\hbar \sin \chi \left[ \cot \theta \dfrac{\partial}{\partial \chi}- \dfrac{1}{\sin\theta}\dfrac{\partial}{\partial \phi} \right]- -i\hbar \cos\chi \dfrac{\partial}{\partial \theta} \]
\[ J_c = -i\hbar\dfrac{\partial}{\partial \chi}. \]
Los ángulos\(\theta, \phi, \text{ and } \chi\) son los ángulos de Euler necesarios para especificar la orientación de la molécula rígida en relación con un sistema de coordenadas fijado en laboratorio. El cuadrado correspondiente del operador de momento angular total se\(J^2\) puede obtener como
\[ J^2 = J_a^2 + J_b^2 + J_c^2 = -\dfrac{\partial^2}{\partial\theta^2} - cot\theta \dfrac{\partial}{\partial \theta} - \dfrac{1}{sin\theta}\left( \dfrac{\partial^2}{\partial\phi^2} + \dfrac{\partial^2}{\partial\chi^2} - 2cos\theta \dfrac{\partial^2}{\partial\phi\partial\chi}\right), \]
y el componente a lo largo del eje Z fijo en laboratorio\(J_Z \text{ is } -i\hbar\frac{\partial}{\partial \phi}.\)
Las funciones propias y los valores propios para casos especiales
Tops esféricos
Cuando los tres valores principales del momento de inercia son idénticos, la molécula se denomina una parte superior esférica. En este caso, la energía rotacional total se puede expresar en términos del operador de momento angular total\(J^2\)
\[ H_{rot} = \dfrac{J^2}{2I} \]
Como resultado, las funciones propias de\(H_{rot}\) son las de\(J^2\) (y\(J_a \text{ as well as }J_Z \text{ both of which commute with }J^2 \text{ and with one another; } J_Z \) es el componente de J a lo largo del eje Z fijo en laboratorio y los desplazamientos con\(J_a \text{ because } J_Z = - i\hbar\frac{\partial}{\partial \phi} \text{ and } J_a = -i\hbar\frac{\partial}{\partial\chi}\) actúan en diferentes ángulos). Las energías asociadas con tales funciones propias son
\[ E(J,M,K) = \hbar^2\dfrac{J(J+1)}{2I^2}, \]
para todos los K (es decir, números\(J_a\) cuánticos) que van de -J a J en pasos unitarios y para todos los M (es decir, números\(J_Z\) cuánticos) que van de -J a J. Cada nivel de energía es por lo tanto\((2J + 1)^2\) degenarado porque hay 2J + 1 posibles valores K y 2J + 1 posibles valores M para cada J.
Las funciones propias de\(J^2, J_Z \text{ and } J_a , |J,M,K \rangle\) se dan en términos del conjunto de matrices de rotación\(D_{J,M,K}:\)
\[ |J,M,K \rangle = \sqrt{\dfrac{2J + 1}{8\pi^2}}D^{\text{*}}_{J,M,K}(\theta ,\phi ,\chi) \]
que obedecen
\[ J^2|J,M,K \rangle = \hbar^2 J(J+1) |J,M,K\rangle, \]
\[ J_a|J,M,K\rangle = \hbar K|J,M,K\rangle, \]
\[ J_Z|J,M,K\rangle = \hbar M|J,M,K\rangle. \]
Tops simétricos
Las moléculas para las que dos de los tres momentos principales de inercia son iguales se denominan cimas simétricas. Aquellos para los que el momento único de inercia es menor que los otros dos se denominan cimas simétricas prolongadas; si el momento único de inercia es mayor que los otros, la molécula es una parte superior simétrica oblata. Nuevamente, la energía cinética rotacional, que es la hamiltoniana rotacional completa, se puede escribir en términos del operador de momento angular rotacional total\(J^2\) y la componente del momento angular a lo largo del eje con el único momento principal de inercia:
\[ H_{rot} = \dfrac{J^2}{2I} + J_a^2\left[ \dfrac{1}{2I_a} - \dfrac{1}{2I} \right]\]
para las tapas proladas y
\[ H_{rot} = \dfrac{J^2}{2I} + J_c^2\left[ \dfrac{1}{2I_c} - \dfrac{1}{2I}\right]\]
para tops oblatos.
Como resultado, las funciones propias de\(H_{rot} \text{are those of }J^2 \text{ and } J_a \text{ or }J_c \text{ (and of }J_Z)\), y los niveles de energía correspondientes son:
\[ E(J,K,M) = \hbar^2\dfrac{J(J+1)}{2I^2} + \hbar^2K^2\left[ \dfrac{1}{2I_a} - \dfrac{1}{2I} \right], \]
para las toops proladas
\[ E(J,K,M) = \hbar \dfrac{J(J+1)}{2I^2} + \hbar^2K^2 \left[ \dfrac{1}{2I_a} - \dfrac{1}{2I} \right], \]
para tops oblatos, nuevamente para K y M (es decir, números\(J_a \text{ or } J_c \text{ and } J_Z\) cuánticos, respectivamente) que van de -J a J en pasos unitarios. Dado que la energía ahora depende de K, estos niveles son solo 2J + 1 degenerados debido a los diferentes valores de M de 2J + 1 que surgen por cada valor de J. \(|J, M,K \rangle\)Las funciones propias son las mismas funciones de matriz de rotación que surgen para el caso esférico superior.
Tops Asimétricos
Las funciones propias rotacionales y los niveles de energía de una molécula para la que los tres momentos principales de inercia son distintos (una llamada parte superior asimétrica) no se pueden expresar fácilmente en términos de los estados propios de momento angular y los números cuánticos J, M y K. Sin embargo, dados los tres momentos principales de inercia\(I_a, I_b, \text{ and } I_c\), una representación matricial de cada una de las tres contribuciones a la rotación hamiltoniana
\[ H_{rot} = \dfrac{J_a^2}{2I_a} + \dfrac{J_b^2}{2I_b} + \dfrac{J_c^2}{2I_c} \]
se puede formar dentro de un conjunto de bases de las funciones de matriz de rotación {|J, M, K>}. Esta matriz no será diagonal porque las funciones |J, M, K> no son funciones propias de la parte superior asimétrica\(H_{rot}\). Sin embargo, la matriz se puede formar en esta base y posteriormente llevarla a forma diagonal al encontrar sus vectores propios {\(C_{n,J,M,K}\)y sus valores propios\(\{E_n\}\). Los coeficientes vectoriales expresan los estados propios asimétricos superiores como
\[ \Psi_n(\theta , \phi , \chi ) = \sum\limits_{J, M, K} C_{n, J, M, K}|J, M, K\rangle. \]
Debido a que el momento angular total\(J^2\) aún se desplaza\(H_{rot}\), cada uno de esos autoestados contendrá solo un valor J y, por lo tanto, también se\(\Psi_n\) puede etiquetar con un número cuántico J:
\[ \Psi_{n,J}(\theta , \phi, \chi) = \sum\limits_{M,K} C_{n, J, M, K} |J, M, K\rangle. \]
Para formar los únicos elementos de matriz distintos de cero\(H_{rot}\) dentro de la\(|J, M, K \rangle\) base, se pueden usar las siguientes propiedades de las funciones rotación-matriz:
\[ \langle J, M, K| J_a^2 | J, M, K \rangle =\langle J, M, K| J_b^2 |J, M, K \rangle = \dfrac{1}{2} \langle J,M, K| J^2 - J_c^2 | J, M, K \rangle = \hbar^2 [J(J+1) - K^2], \]
\[ \langle J, M, K | J_c^2 |J, M, K \rangle = \hbar^2 K^2, \]
\[ \langle J, M, K| J_a^2 | J, M, K \pm 2 \rangle = -\langle J, M, K | J_b^2 | J, M, K \pm 2 \rangle = \hbar^2 \sqrt{J(J+1) - K(K\pm 1}\sqrt{J(J+1) - (K \pm 1)(K \pm 2)} \]
\[ \langle J, M, K| J_c^2 | J, M, K \pm 2 \rangle = 0. \]
Cada uno de los elementos del\(J_c^2, J_a^2, \text{ and }J_b^2\) mosto, por supuesto, ser multiplicado, respectivamente, por\( \frac{1}{2I_c}, \frac{1}{2I_a} \text{ and } \frac{1}{2I_b}\) sumados juntos para formar la representación matricial de\(H_{rot}.\) La diagonalización de esta matriz proporciona entonces las energías superiores asimétricas y las funciones de onda.
Colaboradores y Atribuciones
Jack Simons (Henry Eyring Scientist and Professor of Chemistry, U. Utah) Telluride Schools on Theoretical Chemistry and Jeff A. Nichols (Oak Ridge National Laboratory)