
13.2: Movimiento vibratorio dentro de la aproximación armónica
- Page ID
- 74366

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
El simple movimiento armónico de una molécula diatómica fue tratado en el Capítulo 1, y no se repetirá aquí. En cambio, se pone énfasis en las moléculas poliatómicas cuya dependencia de la energía electrónica de las coordenadas cartesianas 3N de sus átomos de N puede escribirse (aproximadamente) en términos de una expansión de la serie Taylor alrededor de un mínimo local estable. Por lo tanto, asumimos que la molécula de interés existe en un estado electrónico para el cual la geometría que se considera es estable (es decir, no sujeta a distorsión geométrica espontánea).
La expansión de la serie Taylor de la energía electrónica está escrita como:
\[ V(q_k) = V(0) + \sum\limits_k \left( \dfrac{\partial V}{\partial q_k} \right) q_k + \dfrac{1}{2}\sum\limits_{j,k}q_j H_{j,k} + ... , \]
donde V (0) es el valor de la energía electrónica en la geometría estable en estudio,\(q_k\) es el desplazamiento de la coordenada\(k_{th}\) cartesiana alejándose de esta posición inicial,\(\left( \frac{\partial V}{\partial q_k} \right)\) es el gradiente de la energía electrónica a lo largo de esta dirección, y los\( H_{j,k} \) son los segundos elementos de matriz derivada o hessiana a lo largo de estas direcciones\(H_{j,k} = \left( \frac{\partial^2 V}{\partial q_j\partial q_k} \right).\) Si la geometría inicial corresponde a una especie estable, los términos de gradiente desaparecerán (es decir, esta geometría corresponde a un punto mínimo, máximo o de sillín), y la matriz de hessian poseerá 3N - 5 (para lineal especies) o 3N -6 (para moléculas no lineales) valores propios positivos y 5 o 6 valores propios cero (correspondientes a 3 movimientos de traducción y 2 o 3 movimientos rotacionales de la molécula). Si el hessian tiene un valor propio negativo, la geometría corresponde a un estado de transición (estas situaciones se discuten en detalle en el Capítulo 20).
A partir de ahora, asumimos que la geometría en estudio corresponde a la de un mínimo estable sobre el cual se produce el movimiento vibratorio. El tratamiento de geometrías inestables es de gran importancia para la química, pero este Capítulo trata sobre las vibraciones de especies estables. Para un buen tratamiento de situaciones bajo las cuales se espera que ocurra inestabilidad geométrica, véase el Capítulo 2 del texto Principios Energéticos de las Reacciones Químicas de J. Simons. En el Capítulo 20 del presente texto se ofrece una discusión sobre cómo se ubican los mínimos locales y los estados de transición en las superficies de energía electrónica.
Las ecuaciones de Newton del movimiento para la vibración
Las matrices de energía cinética y potencial
Truncando la serie Taylor en términos cuadráticos (asumiendo que estos términos dominan porque solo pequeños desplazamientos de la geometría de equilibrio son de interés), uno tiene el llamado potencial armónico:
\[ V(q_k) = V(0) + \dfrac{1}{2}\sum\limits_{j,k}q_j H_{j,k}q_k. \]
Las ecuaciones mecánicas clásicas de movimiento para las coordenadas 3N {\(q_k\)} se pueden escribir en términos de la energía potencial anterior y la siguiente función de energía cinética:
\[ T = \dfrac{1}{2}\sum\limits_jm_j\dot{q_j}^2, \]
donde\(\dot{q_j}\) denota la tasa de tiempo de cambio de la coordenada\(q_j \text{ and } m_j\) es la masa del átomo en el que reside la coordenada\(j^{th}\) cartesiana. Las ecuaciones de Newton así obtenidas son:
\[ m_j \ddot{q_j} = - \sum\limits_k H_{j,k}q_k \]
donde la fuerza a lo largo de la\(j^{th}\) coordenada viene dada por menos la derivada del potencial V a lo largo de esta coordenada\( \frac{\partial V}{\partial q_j}= \sum\limits_k H_{j,k}q_k \) dentro de la aproximación armónica.
Estas ecuaciones clásicas pueden expresarse de manera más compacta en términos de la evolución temporal de un conjunto de las llamadas coordenadas cartesianas ponderadas en masa definidas como:
\[ x_j = q_j\sqrt{m_j}, \]
en términos de los cuales las ecuaciones de Newton se convierten
\[ \ddot{x_j} = -\sum\limits_k H'_{j,k}x_k \]
y los elementos de la matriz de hessian ponderados en masa son
\[ H'_{j,k} = H_{j,k}\dfrac{1}{\sqrt{m_jm_k}}. \]
Las energías vibracionales armónicas y los vectores propios de modo normal
Asumiendo que se\(x_j\) someten a alguna forma de evolución del tiempo sinusoidal:
\[ x_j(t) = x_j(0)cos(\omega t), \]
y sustituyendo esto en las ecuaciones de Newton produce una ecuación de valor propio de matriz:
\[ \omega^2x_j = \sum\limits_k H'_{j,k}x_k \]
en el que los autovalores son los cuadrados de las llamadas frecuencias vibracionales de modo normal y los vectores propios dan las amplitudes de movimiento a lo largo de cada una de las coordenadas cartesianas ponderadas de masa 3N que pertenecen a cada modo.
Dentro de este tratamiento armónico del movimiento vibratorio, la energía vibratoria total de la molécula se da como
\[ E(v_1, v_2, ... V_{3N-5 \text{ or }6}) = \sum\limits_{j=1}^{3N-5 \text{ or } 6} \hbar\omega_j \left( v_j + \dfrac{1}{2} \right) \]
como producto de las funciones del oscilador armónico 3N-5 o 3N-6\(\Psi_{vj}(x_j)\) son para cada modo normal dentro de esta imagen, la brecha de energía entre un nivel vibracional y otro en el que uno de los números\(v_j\) cuánticos se incrementa por unidad (el origen de esta “regla de selección” se discute en el Capítulo 15) es
\[ \Delta E_{vj} \rightarrow v_j + 1 = \hbar\omega_j \]
Por lo tanto, el modelo armónico predice que la\((v=0 \rightarrow v = 1) \text{ and "hot band" } (v=1 \rightarrow v = 2)\) transición “fundamental” debe ocurrir con la misma energía, y las transiciones armónicas (v=0 Æ v=2) deberían ocurrir exactamente al doble de esta energía.
El uso de la simetría
Modos adaptados a simetría
A menudo es posible simplificar el cálculo de las frecuencias de modo normal y los vectores propios mediante la explotación de la simetría del grupo de puntos moleculares. Para las moléculas que poseen simetría, el potencial electrónico\(V(q_j)\) muestra simetría con respecto a desplazamientos de simetría equivalente a coordenadas cartesianas. Por ejemplo, considere la molécula de agua en su geometría de\(C_{2v}\) equilibrio como se ilustra en la siguiente figura. Un movimiento muy pequeño del átomo H izquierdo de la\(H_2O\) molécula en la dirección x positiva\((\Delta x_L)\) produce el mismo cambio en V que un desplazamiento correspondientemente pequeño del átomo H derecho en la dirección x negativa.\((-\Delta x_R).\) De manera similar, el movimiento de la H izquierda en la dirección y positiva\((\Delta y_L)\) produce un cambio de energía idéntico al movimiento de la derecha H en la dirección y positiva\((\Delta y_R).\)
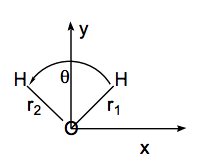
La equivalencia de los pares de desplazamientos de coordenadas cartesianas es el resultado de que los vectores de desplazamiento están conectados por las operaciones de grupo de puntos del\(C_{2v}\) grupo. En particular, la reflexión de\(\Delta x_L\) a través del plano yz produce\(-\Delta x_R\), y la reflexión de\(\Delta y_L\) a través de este mismo plano produce\(\Delta y_R.\)
De manera más general, es posible combinar conjuntos de coordenadas de desplazamiento cartesiano {\(q_k\)} en las llamadas coordenadas adaptadas a simetría {\(Q_{Gamma,j}\)}, donde el índice\(\Gamma\) etiqueta la representación irreducible y j etiqueta la combinación particular de esa simetría. Estas coordenadas adaptadas a simetría se pueden formar aplicando los operadores de proyección de grupo de puntos a las coordenadas individuales de desplazamiento cartesiano.
Para ilustrar, nuevamente considere la\(H_2O\) molécula en el sistema de coordenadas descrito anteriormente. Las coordenadas de desplazamiento cartesiano ponderado en masa 3N = 9\((X_L, Y_L, Z_L, X_O, Y_O, Z_O, X_R, Y_R, Z_R)\) pueden adaptarse a la simetría aplicando los siguientes cuatro operadores de proyección:
\[ P_{A_1} = 1 + \sigma_{yz} + \sigma_{xy} + C_2 \]
\[ P_{b_1} = 1 + \sigma_{yz} + \sigma_{xy} - C_2 \]
\[ P_{b_2} = 1 - \sigma_{yz} + \sigma_{xy} - C_2 \]
\[ P_{a_2} = 1 - \sigma_{yz} - \sigma_{xy} + C_2 \]
a cada una de las 9 coordenadas originales. Por supuesto, no se obtendrán 9 x 4 = 36 coordenadas independientes adaptadas a simetría de esta manera; surgirán muchas combinaciones idénticas, y solo 9 serán independientes.
La combinación independiente de\ (\ textbf {a_1 simetría} (normalizada para producir vectores de longitud unitaria) son
\[ Q_{a_1,1} = \dfrac{1}{\sqrt{2}}[X_L - X_R] \]
\[ Q_{a_1,2} = \dfrac{1}{\sqrt{2}}[Y_L - Y_R] \]
\[ Q_{a_1,3} = [Y_O] \]
Los de\(b_2\) simetría son
\[ Q_{b_2,1} = \dfrac{1}{\sqrt{2}}[X_L + X_R] \]
\[ Q_{b_2,2} = \dfrac{1}{\sqrt{2}}[Y_L - Y_R] \]
\[ Q_{b_2,3} = [X_O] \]
y las combinaciones
\[ Q_{b_1,1} = \dfrac{1}{\sqrt{2}}[Z_L + Z_R] \]
\[ Q_{b_1,2} = [Z_O] \]
son de\( b_1 \) simetría, mientras
\[ Q_{a_2,1} = \dfrac{1}{\sqrt{2}}[Z_L - Z_R] \]
es de\(a_2\) simetría.
Grupo de puntos Simetría del Potencial Armónico
Estos nueve\(Q_{\Gamma,j}\) se expresan como transformaciones unitarias de las coordenadas cartessianas ponderadas en masa originales:
\[ Q_{\Gamma,j} = \sum\limits_k C_{\Gamma, j, k}X_k \]
Estos coeficientes de transformación {\(C_{\Gamma, j, k}\)} pueden ser utilizados para llevar a cabo una transformación unitaria de la matriz de hessiana ponderada en masa 9x9. Al hacerlo, solo necesitamos formar bloques
\[ H^{\Gamma}_{j,1} = _{k k'}C_{\Gamma , j, k}H_{k, k'}\dfrac{1}{\sqrt{m_k m_k'}}C_{\Gamma , 1, k'} \]
dentro del cual las simetrías de los dos modos son idénticas. Los elementos fuera de la diagonal
\[ H_{j 1}^{\Gamma \Gamma '} = _{k k'}C_{\Gamma , j, k}H_{k, k'}\dfrac{1}{\sqrt{m_km_k'}}C_{\Gamma ' , 1, k'} \]
desaparecer porque las operaciones potenciales de simetría del grupo de\(V(q_j) \text{ (and the full vibrational Hamiltonian H = T + V) commutes with the }C_{2V}\) puntos.
Como resultado, el problema del valor propio hessiano ponderado en masa 9x9 se puede subdividir en dos problemas de matriz 3x3 (de\(a_1 \text{ and } b_2\) simetría), una matriz 2x2 de\(b_1 \text{ symmetry and one 1x1 matrix of } a_2 \text{ symmetry. For example, the } a_1 \text{ symmetry block } H^{a_1}_{ j l}\) se forma de la siguiente manera:
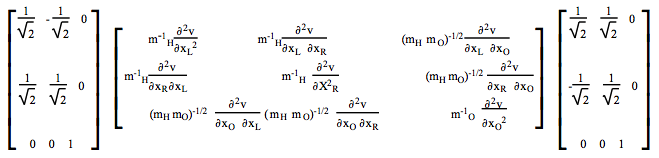
Los\(b_2, b_1 \text{ and } a_2\) bloques se forman de manera similar. Los valores propios de cada uno de estos bloques proporcionan los cuadrados de las frecuencias vibracionales armónicas, los vectores propios proporcionan los desplazamientos de modo normal como combinaciones lineales de la simetría adaptada {\(Q^{\Gamma}_{j}\)}.
Independientemente de si se usa simetría para bloquear diagonalizar el hessian ponderado en masa, seis (para moléculas no lineales) o cinco (para especies lineales) de los valores propios serán iguales a cero. Los vectores propios que pertenecen a estos valores propios cero describen las 3 traducciones y 2 o 3 rotaciones de la molécula. Por ejemplo,
\[ \dfrac{1}{\sqrt{3}}[X_L + X_R + X_O] \]
\[ \dfrac{1}{\sqrt{3}}[Y_L + Y_R + Y_O] \]
\[ \dfrac{1}{\sqrt{3}}[Z_L + Z_R + Z_O] \]
son tres vectores propios de traducción de\(b_2, a_1 \text{ and } b_1\) simetría, y
\[ \dfrac{1}{\sqrt{2}}(Z_L - Z_R) \]
es una rotación (alrededor del eje Y en la figura mostrada anteriormente) de\(a_2\) simetría. Este vector de rotación se puede generar aplicando el\(a_2 \text{ projection operator to } Z_L \text{ or to } Z_R.\) El hecho de que la rotación alrededor del eje Y sea de\(a_2 \text{ symmetry is indicated in the right-hand column of the } C_{2v} \text{ character table of Appendix E via the symbol } R_Z\) (n.b., se debe tener cuidado en darse cuenta de que la convención del eje utilizada en la figura anterior es diferente a la implícita en la tabla de caracteres; esta última tiene el eje Z fuera del molecular plano, mientras que la figura llama a esto el eje X). Las otras dos rotaciones son de tabla de\(b_1 \text{ and } b_2 \text{ symmetry (see the } C_{2v}\) caracteres del Apéndice E) e implican el giro de la molécula alrededor de los ejes X y Z de la figura dibujada anteriormente, respectivamente.
Entonces, de los 9 desplazamientos cartesianos, 3 son\(a_1 \text{ symmetry, 3 of } b_2 ,\text{ 2 of } b_1,\text{ and 1 of } a_2.\) de De estos, hay tres traslaciones\((a_1, b_2,\text{ and } b_1)\) y tres rotaciones\((b_2, b_1,\text{ and } a_2).\) Esto deja dos vibraciones de\(a_1 \text{ and one of } b_2\) simetría. Para el\(H_2O\) ejemplo tratado aquí, los tres valores propios distintos de cero del hessian ponderado en masa son, por lo tanto, de\(a_1 b_2 ,\text{ and } a_1\) simetría. Describen las vibraciones de estiramiento simétricas y asimétricas y el modo de flexión, respectivamente, como se ilustra a continuación.
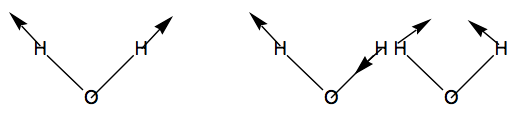
El método de análisis vibracional presentado aquí puede funcionar para cualquier molécula poliatómica. Se conoce el hessian ponderado en masa y luego calcula los valores propios distintos de cero que luego proporcionan los cuadrados de las frecuencias vibracionales de modo normal. La simetría de grupo de puntos se puede utilizar para bloquear diagonalizar este hessian y etiquetar los modos vibracionales de acuerdo a la simetría.
Colaboradores y Atribuciones
Jack Simons (Henry Eyring Scientist and Professor of Chemistry, U. Utah) Telluride Schools on Theoretical Chemistry and Jeff A. Nichols (Oak Ridge National Laboratory)