
18.4: Conjuntos de Bases Orbitales Atómicas
- Page ID
- 74626

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Los orbitales de base comúnmente utilizados en el proceso LCAO-MO-SCF se clasifican en dos clases orbitales tipo Slater y orbitales cartesianos de tipo gaussiano.
Los orbitales tipo Slater-type (STO) se caracterizan por números cuánticos\(n\)\(l\),\(m\) y y exponentes (que caracterizan el 'tamaño' de la función base)\(\xi\):
\[ \chi_{n,l,m}(r,\theta ,\phi )= N_{n,l,m,\xi} Y_{l,m}(\theta ,\phi )r^{n-1}e^{-\xi r}, \]
y\(N_{n,l,m, \xi}\) denota la constante de normalización.
Los orbitales cartesianos de tipo gaussiano (GTO) se caracterizan por números cuánticos\(a\)\(b\), y\(c\) que detallan la forma angular y la dirección de la órbita y los exponentes\(\alpha\) que gobiernan el “tamaño” radial de la función base similar a\(\xi\) los STOs.
\[ \chi_{a,b,c}(r, \theta ,\phi )- N'_{a,b,c,\alpha}\textbf{x}^a\textbf{y}^b\textbf{z}^ce^{-\alpha r^2}, \]
Por ejemplo, orbitales con\(a\)\(b\), y\(c\) valores de 1,0,0 o 0,1,0 o 0,0,1 son\(p_x\),\(p_y\), y\(p_z\) orbitales; aquellos con\(a\), y\(c\) valores de 2\(b\), 0, 0 o 0, 2, 0 o 0, 0, 0 o 0, 0, 2 y 1, 0 o 0, 1, 1 o 1, 0, 1 abarcan el espacio de cinco d orbitales y orbitales one s (la suma de los orbitales 2, 0, 0 y 0, 2, 0 y 0, 0, 2 es un orbital s porque\(x^2 + y^2 + z^2 = r^2\) es independiente de\(\theta \text{ and } \phi \)).
Para ambos tipos de orbitales, las coordenadas\(r\, \theta, \text{ and } \phi\) se refieren a la posición del electrón con respecto a un conjunto de ejes unidos al centro sobre el que se ubica la base orbital.
La utilidad de los gaussianos
Aunque las STO son preferidas por razones fundamentales (por ejemplo, como se demuestra en los Apéndices A y B, los orbitales de átomos de hidrógeno son de esta forma y la solución exacta de la ecuación de Schrödinger de muchos electrones puede mostrarse que es de esta forma (en cada una de sus coordenadas) cerca de los centros nucleares), se utilizan STO principalmente para cálculos atómicos y de moléculas lineales porque las integrales multicéntricas\( \langle \chi_a \chi_b \big| g \big| \chi_e \chi_d \rangle \) (cada base orbital puede estar en un centro atómico separado) que surgen en los cálculos de moléculas poliatómicas no pueden realizarse de manera eficiente cuando se emplean STO. Por el contrario, tales integrales se pueden hacer rutinariamente cuando se usan GTO. Esta ventaja fundamental de los GTO ha llevado al dominio de estas funciones en la química cuántica molecular.
Para entender por qué las integrales sobre los GTO pueden llevarse a cabo cuando las integrales análogas basadas en STO son mucho más difíciles, solo se deben considerar los productos orbitales (\( \chi_a \chi_c (r_1) \text{ and } \chi_b \chi_d (r_2) \)) que surgen en tales integrales. Para los orbitales de la forma GTO, dichos productos involucran\( e^{-\alpha_a (\textbf{r}-\textbf{R}_a)^2}e^{-\alpha_c( \textbf{r}-\textbf{R}_c )^2} \). Al completar el cuadrado en el exponente, este producto se puede reescribir de la siguiente manera:
\[ e^{-\alpha_a (\textbf{r}-\textbf{R}_a)^2} e^{-\alpha_c (\textbf{r}-\textbf{R}_c)^2} = e^{-(\alpha_a + \alpha_c )(\textbf{r}-\textbf{R'})^2}e^{-\alpha ' (\textbf{R}_a-\textbf{R}_c)^2}, \]
donde
\[ \textbf{R}' = \dfrac{\alpha_a\textbf{R}_a + \alpha_c\textbf{R}_c}{\alpha_a + \alpha_c}\]
y
\[ \alpha ' = \dfrac{\alpha_a \alpha_c}{\alpha_a + \alpha_c}. \]
Así, el producto de dos GTO en diferentes centros es igual a otro GTO único en un centro\(\textbf{R}'\) entre los dos centros originales. Como resultado, incluso una integral de dos electrones de cuatro centros sobre GTO se puede escribir como, a lo sumo, una integral de dos electrones de dos centros; resulta que esta reducción en los centros es suficiente para permitir que se lleven a cabo todas esas integrales. No se presenta una reducción similar para las STOs porque el producto de dos STOs no se puede reescribir como una nueva STO en un nuevo centro.
Para superar la debilidad primaria de las funciones de GTO, que tienen un comportamiento incorrecto cerca de los centros nucleares (es decir, sus derivadas radiales desaparecen en el núcleo mientras que las derivadas de las STO son distintas de cero), es común combinar dos, tres o más GTO, con coeficientes de combinación que son fijos y no tratados como parámetros LCAO-MO, en nuevas funciones llamadas GTO contratados o CGTO. Típicamente, una serie de GTO ajustados, medios y sueltos (es decir, GTO con\(\alpha\) valores grandes, medianos y pequeños, respectivamente) se multiplican por los llamados coeficientes de contracción y se suman para producir un CGTO que parece poseer la 'cúspide' apropiada (es decir, pendiente distinta de cero) en el centro nuclear (aunque incluso tal combinación no puede porque cada GTO tiene pendiente cero en el núcleo).
Bibliotecas Basis Set
Se ha dedicado mucho esfuerzo al desarrollo de conjuntos de orbitales de base STO o GTO para los elementos del grupo principal y los metales de transición más ligeros. Este esfuerzo continuo está dirigido a proporcionar bibliotecas de conjunto de bases estándar que:
- Rendimiento de precisión química razonable en las funciones de onda y energías resultantes.
- Son rentables ya que su uso en cálculos prácticos es factible.
- Son relativamente transferibles en el sentido de que la base de un átomo dado es lo suficientemente flexible como para ser utilizada para ese átomo en una variedad de entornos de unión (donde la hibridación del átomo y la polaridad local pueden variar).
El núcleo fundamental y la base de valencia
Al construir una base orbital atómica para usar en un cálculo particular, se debe elegir entre varias clases de funciones. Primero, se debe especificar el tamaño y la naturaleza del núcleo primario y la base de valencia. Dentro de esta categoría, son comunes las siguientes opciones:
- Una base mínima en la que el número de orbitales STO o CGTO es igual al número de orbitales atómicos de núcleo y valencia en el átomo.
- Una base de doble zeta (DZ) en la que se utilizan el doble de STOs o CGTO que los orbitales atómicos de núcleo y valencia. El uso de más funciones básicas está motivado por el deseo de proporcionar flexibilidad variacional adicional al proceso LCAO-MO. Esta flexibilidad permite que el proceso LCAO-MO genere orbitales moleculares de difusión variable a medida que varía la electronegatividad local del átomo. Típicamente, las bases de doble zeta incluyen pares de funciones con un miembro de cada par que tiene un exponente (\(\zeta \text{ or } \alpha\)valor) más pequeño que en la base mínima y el otro miembro tiene un exponente mayor.
- Una base triple zeta (TZ) en la que se utilizan tres veces más STO o CGTO que el número de orbitales atómicos de núcleo y valencia.
- Dunning ha desarrollado bases CGTO que van desde aproximadamente DZ hasta sustancialmente más allá de la calidad TZ (T. H. Dunning, J. Chem. Phys. 53, 2823 (1970); T. H. Dunning y P. J. Hay en Métodos de Teoría de la Estructura Electrónica, H. F. Schaefer, III Ed., Plenum Press, Nueva York (1977)). Estas bases implican contracciones de bases primitivas GTO que Huzinaga había optimizado anteriormente (S. Huzinaga, J. Chem. Phys. 42, 1293 (1965)) para su uso como funciones no contratadas (es decir, para las cuales Huzinaga varió los\(\alpha\) valores para minimizar las energías de varios estados electrónicos del átomo correspondiente). Estas bases Dunning se denotan comúnmente, por ejemplo, de la siguiente manera para los átomos de la primera fila: (10s,6p/5s,4p), lo que significa que se han contraído 10 GTO primitivos de tipo s para producir 5 CGTOs de tipo s separados y que 6 GTO primitivos de tipo p se contrajeron para generar 4 CGTOs de tipo p separados. Los conjuntos de bases más recientes del grupo Dunning se dan en T. Dunning, J. Chem. Phys. 90, 1007 (1990).
- Juegos de bases de templado uniforme (M. W. Schmidt y K. Ruedenberg, J. Chem. Phys. 71, 3961 (1979)) consisten en GTO en los que los exponentes orbitales ak pertenecientes a series de orbitales consisten en progresiones geométricas:\(\alpha_k \text{ = a } \beta_k\), dónde\(\alpha\) y\(\beta\) caracterizan el conjunto particular de GTO.
- Las bases STO-3G se emplearon hace algunos años (W. J. Hehre, R. F. Stewart, y J. A. Pople, J. Chem. Phys. 51, 2657 (1969)) pero son menos populares recientemente. Estas bases están construidas por mínimos cuadrados ajustando GTO a STOs que han sido optimizados para diversos estados electrónicos del átomo. Cuando se emplean tres GTO para adaptarse a cada STO, se forma una base STO-3G.
- Bases 4-31G, 5-31G y 6-31G (R. Ditchfield, W. J. Hehre, y J. A. Pople, J. Chem. Phys. 54, 724 (1971); W. J. Hehre, R. Ditchfield, y J. A. Pople, J. Chem. Phys. 56, 2257 (1972); P. C. Hariharan y J. A. Pople, Teoreta. Chim. Acta. (Berl.) 28, 213 (1973); R. Krishnan, J. S. Binkley, R. Seeger, y J. A. Pople, J. Chem. Phys. 72, 650 (1980)) emplean un solo CGTO de longitud de contracción 4, 5 o 6 para describir el núcleo orbital. El espacio de valencia se describe en el nivel DZ con el primer CGTO construido a partir de 3 GTO primitivos y el segundo CGTO construido a partir de un solo GTO primitivo.
Los valores de los exponentes orbitales (\(\zeta s \text{ or } \alpha s\)) y los coeficientes de contracción Gto-to-CGTO necesarios para implementar una base particular del tipo descrito anteriormente han sido tabulados en varios artículos de revistas y en bases de datos informáticas (en particular, en la base de datos contenida en el libro Handbook of Conjuntos de bases gaussianas: A. Compendio para Ab initio Cálculos Orbitales Moleculares, R. Poirer, R. Kari, e I. G. Csizmadia, Elsevier Science Publishing Co., Inc., Nueva York, Nueva York (1985)).
Varias otras fuentes de conjuntos de bases para átomos particulares se enumeran en la Tabla que se muestra a continuación (aquí JCP y JACS son abreviaturas para el Journal of Chemical Physics y el Journal of The American Chemical Society, respectivamente).
| Referencia de la literatura | Tipo de Base | Átomos |
|---|---|---|
| Hehre, W.J.; Stewart, R.F.; Pople, J.A. JCP 51, 2657 (1969). Hehre, W.J.; Ditchfield, R.; Stewart, R.F.; Pople, J.A. JCP 52, 2769 (1970). |
STO - 3G | H-Ar |
| Binkley, J.S.; Pople, J.A.; Hehre, W.J. JACS 102, 939 (1980). | 3-21G | H-Ne |
| Gordon, M.S.; Binkley, J.S.; Pople, J.A.; Pietro, W.J.; Hehre, W.J. JACS 104, 2797 (1982). | 3-21G | Na-Ar |
| Dobbs, K.D.; Hehre, W.J., J. Comput. Chem. 7, 359 (1986). | 3-21G | K, Ca, Ga |
| Dobbs, K.D.; Hehre, W.J., J. Comput. Chem. 8, 880 (1987). | 3-21G | Sc-Zn |
| Ditchfield, R.; Hehre, W.J.; Pople, J.A., JCP 54, 724 (1971). | 6-31G | H |
| Eneldo, J.D.; Pople, J.A., JCP 62, 2921 (1975). | 6-31G | Li, B |
| Binkley, J.S.; Pople, J.A., JCP 66, 879 (1977) | 6-31G | Be |
| Hehre, W.J.; Ditchfield, R.; Pople, J.A., JCP 56, 2257 (1972). | 6-31G | C-F |
| Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. JCP 77, 3654 (1982). |
6-31G | Na-Ar |
| Dunning, T. JCP 53, 2823 (1970). | (4s/2s) (4s/3s) (9s5p/3s2p) (9s5p/4s2p) (9s5p/5s3p) |
H H B-F B-F B-F |
| Dunning, T. JCP 55, 716 (1971). | (5s/3s) (10s/4s) (10s/5s) (10s6p/5s3p) (10s6p/5s4p) |
H Li Be B-Ne B-Ne |
| Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. JCP 72, 650 (1980). | 6-311G | H-Ne |
| Dunning, inédito VDZ. | (4s/2s) (9s5p/3s2) (12s8p/4s3p) |
H Li, Be, C-Ne Na-Ar |
| Dunning, inédito VTZ. | (5s/3s) (6s/3s) (12s6p/4s3p) (17s10p/5s4p) |
H |
| Dunning, inédito VQZ. | (7s/4s) (8s/4s) (16s7p/5s4p) |
H H B-Ne |
| Dunning, T. JCP 90, 1007 (1989). (PvDz, PvTz, PvQZ consistente en correlación) | (4s1p/2s1p) (5s2p1d/3s2p1d) (6s3p1d1f/4s3p2d1f) (9s4p1d/3s2p1d) (10s5p2d1f/4s3p2d1f) (12s6p3d2f1g/5s4p3d2f1g) |
H H B-Ne B-Ne B-Ne |
| Huzinaga, S.; Klobukowski, M.; Tatewaki, H. Can. J. Chem. 63, 1812 (1985). | (14s/2s) (14s9p/2s1p) (16s9p/3s1p) (16s11p/3s2p) |
Li, Be B-Ne Na-Mg Al-Ar |
| Huzinaga, S.; Klobukowski, M. THEOCHEM. 44, 1 (1988). |
(14s10p/2s1p) |
B-Ne Na-Mg Al-Ar K-Ca Sc-Zn Ga |
| McLean, A.D.; Chandler, G.S. JCP 72, 5639 (1980). | (12s8p/4s2p) (12s8p/5s2p) (12s8p/6s4p) (12s9p/6s4p) (12s9p/6s5p) |
Na-Ar, P\(^-\), S\(^-\), Cl\(^-\) Na-Ar, P\(^-\), S\(^-\), Cl\(^-\) Na-Ar, P\(^-\), S\(^-\), Cl\(^-\) Na-Ar, P\(^-\), S\(^-\), Cl\(^-\) Na-Ar, P\(^-\), S\(^-\), Cl \(^-\) |
| Dunning, T.H.Jr.; Hay, P.J. Capítulo 1 en 'Métodos de teoría de la estructura electrónica', Schaefer, H.F.III, Ed., Plenum Press, N.Y., 1977. |
(11s7p/6s4p) | Al-Cl |
| Hood, D.M.; Pitzer, R.M.; Schaefer, H.F.III, JCP 71, 705 (1979). | (14s11p6d/10s8p3d) | Sc-Zn |
| Schmidt, M.W.; Ruedenberg, K. JCP 71, 3951 (1979). (regular de temperamento uniforme) |
([N] s), N=3-10 ([2N] s), N=3-10 ([2N] s), N=3-14 ([2N] s [N] p), N=3-11 ([2N] s [N] p), N=3-13 ([2N] s [N] p), N=4-12 ([2N-6] s [N] p), N=7-15 |
H He Li, Be B, N-Ne C Na, Mg Al-Ar |
Funciones de polarización
Además de la base fundamental de núcleo y valencia descrita anteriormente, se suele agregar a la base un conjunto de las llamadas funciones de polarización. Las funciones de polarización son funciones de un momento angular superior al que aparece en el espacio orbital de valencia del átomo (por ejemplo, funciones d para C, N y O y funciones p para H). Estas funciones de polarización tienen exponentes (\(\zeta \text{ or } \alpha\)) que hacen que sus tamaños radiales sean similares a los tamaños de los orbitales de valencia primaria (es decir, los orbitales p de polarización del átomo H son similares en tamaño al orbital 1s). Por lo tanto, no son orbitales que proporcionen una descripción del orbital de valencia del átomo con un valor l mayor; dichos orbitales de valencia de mayor l serían radialmente más difusos y, por lo tanto, requerirían el uso de STO o GTO con exponentes más pequeños.
El propósito principal de las funciones de polarización es dar flexibilidad angular adicional al proceso LCAO-MO en la formación de los orbitales moleculares de valencia. Esto se ilustra a continuación donde se ve que la polarización d\(_\pi\) orbitales contribuye a la formación del\(\pi \) orbital de enlace de un grupo carbonilo al permitir la polarización del\(_\pi\) orbital p del átomo de carbono hacia la derecha y del\(_\pi\) orbital p del átomo de oxígeno hacia la izquierda.
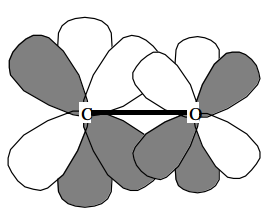
Las funciones de polarización son esenciales en compuestos de anillos tensos porque proporcionan la flexibilidad angular necesaria para dirigir la densidad de electrones hacia regiones entre átomos unidos.
Las funciones con valores l más altos y con 'tamaños' más en línea con los orbitales inferiores también se utilizan para introducir correlación angular adicional en el cálculo permitiendo que se formen pares orbitales polarizados (ver Capítulo 10) que implican correlaciones angulares más altas. Se han tabulado las funciones de polarización óptimas para los átomos de la primera y segunda fila (B. Roos y P. Siegbahn, Teoret. Chim. Acta (Berl.) 17, 199 (1970); M. J. Frisch, J. A. Pople, y J. S. Binkley, J. Chem. Phys. 80, 3265 (1984)).
Funciones difusas
Al tratar con aniones o estados de Rydberg, uno debe aumentar los conjuntos de bases anteriores agregando los llamados orbitales de base difusa. Las funciones convencionales de valencia y polarización descritas anteriormente no proporcionan suficiente flexibilidad radial para describir adecuadamente ninguno de estos casos. Las funciones difusas optimizadas de energía apropiadas para los aniones de la mayoría de los elementos más ligeros del grupo principal se han tabulado en la literatura (una excelente fuente de información del conjunto de bases gaussianas se proporciona en Handbook of Gaussian Basis Sets, R. Poirier, R. Kari, e I. G. Csizmadia, Elsevier, Amsterdam ( 1985)) y en bases de datos. Los conjuntos de bases difusas de Rydberg generalmente se crean agregando a las bases convencionales de valencia-pluspolarización secuencias de GTO primitivos cuyos exponentes son más pequeños que el (llamarlo\(\alpha_{\text{diff}}\)) del GTO más difuso, lo que contribuye fuertemente a las CGTO de valencia. Como “regla general”, se puede generar una serie de tales orbitales difusos que son liniarmente independientes pero que abarcan regiones considerablemente diferentes del espacio radial mediante la introducción de GTO primitivos cuyos exponentes son\( \frac{1}{3}\alpha_{\text{diff}}, \frac{1}{9}\alpha_{\text{diff}}, \frac{1}{27}\alpha_{\text{diff}}, \) etc.
Una vez que se ha especificado una base orbital atómica para cada átomo de la molécula, se puede utilizar el procedimiento LCAO-MO para determinar los\(C_{\nu ,i}\) coeficientes que describen los orbitales ocupados y virtuales en términos del conjunto de bases elegido. Es importante tener en cuenta que los orbitales base no son en sí mismos los verdaderos orbitales de los átomos aislados; incluso los orbitales atómicos propiamente dichos son combinaciones (con valores atómicos para los\(C_{\nu ,i}\) coeficientes) de las funciones base. Por ejemplo, en un tratamiento de nivel básico mínimo del átomo de carbono, el orbital atómico 2s se forma combinando, con signo opuesto para lograr el nodo radial, los dos CGTO (o STOs); la función base de tipo s más difusa tendrá un\(C_{i,\nu}\) coeficiente mayor en el orbital atómico 2s. El orbital atómico 1s se forma combinando los mismos dos CGTO pero con el mismo signo y con la función de base menos difusa teniendo un\(C_{\nu ,i}\) coeficiente mayor. El propio proceso LCAO-MO-SCF determina las magnitudes y signos de la\(C_{\nu ,i}\).
Colaboradores y Atribuciones
Jack Simons (Henry Eyring Scientist and Professor of Chemistry, U. Utah) Telluride Schools on Theoretical Chemistry and Jeff A. Nichols (Oak Ridge National Laboratory)