
8.6: Entalpías Estándar de Reacción
- Page ID
- 74200

El beneficio de estas convenciones es que, a cualquier temperatura particular, el cambio de entalpía estándar para una reacción
\[aA+bB+\dots \ \to \ cC+dD+\dots\nonumber \]
que designamos como\(\Delta_rH^o\), viene dada por
\[\Delta_rH^o=\underbrace{c{\Delta}_fH^o\left(C\right)+d{\Delta}_fH^o\left(D\right)+\dots}_{\text{product enthalpies}} - \underbrace{a{\Delta}_fH^o\left(A\right)-b\Delta_fH^o\left(B\right)-\dots}_{\text{reactant enthalpies}}\nonumber \]
Si tenemos las entalpías de formación, podemos calcular el cambio de entalpía para la reacción. Podemos demostrarlo escribiendo las ecuaciones químicas correspondientes a la formación de A, B, C y D a partir de sus elementos. Cuando multiplicamos estas ecuaciones químicas por el coeficiente estequiométrico debidamente firmado y las sumamos, obtenemos la ecuación química para la reacción indicada de A y B para dar C y D. (Ver abajo.) Debido a que la entalpía es una función de estado, el cambio de entalpía que calculemos de esta manera será válido para cualquier proceso que convierta los reactivos especificados en los productos especificados.
La oxidación del metano a metanol es una reacción que ilustra el valor de este enfoque. Los productos normales en la oxidación del metano son, por supuesto, el dióxido de carbono y el agua. Si la reacción se realiza con un exceso de metano, una porción del producto que contiene carbono será monóxido de carbono en lugar de dióxido de carbono. En cualquier circunstancia, el metanol es, en el mejor de los casos, un producto traza. Sin embargo, sería muy deseable idear un catalizador que convierta de manera cuantitativa o casi cuantitativa el metano en metanol de acuerdo con la ecuación
\[\ce{CH4 + 1/2O_2 -> CH3OH}\nonumber \]
(Esto se denomina frecuentemente una oxidación selectiva, para distinguirla de la oxidación no selectiva que produce dióxido de carbono y agua).
Si el catalizador no fuera desmesuradamente caro o de corta duración, y la presión de operación fuera suficientemente baja, este sería un método económico para la fabricación de metanol. (El metanol se fabrica actualmente a partir de metano. Sin embargo, el proceso implica dos pasos y requiere una inversión sustancial de capital.) Si se pudiera disminuir suficientemente el costo de fabricación del metanol, sería económicamente factible convertir el gas natural, que no puede transportarse económicamente a menos que sea factible construir una tubería para ese propósito, en metanol líquido, que es fácilmente transportado por barco. (En la actualidad, la viabilidad económica del transporte marítimo de gas natural licuado, GNL, es marginal, pero parece estar mejorando). Esta tecnología permitiría utilizar el valor combustible de los recursos conocidos de gas natural que actualmente son inútiles porque están ubicados demasiado lejos de los centros de población.
Cuando contemplamos tratar de desarrollar un catalizador y una planta de fabricación para llevar a cabo esta reacción, pronto descubrimos razones para querer conocer el cambio de entalpía. Una es que la fabricación oxidativa del metanol será exotérmica, por lo que quemar el metanol producido producirá menos calor del que se produciría quemando el metano del que se produjo. Queremos saber cuánta energía térmica se pierde de esta manera.
Otra razón es que una planta de fabricación tendrá que controlar la temperatura de la reacción de oxidación para mantener un rendimiento óptimo. (Si la temperatura es demasiado baja, la velocidad de reacción será demasiado lenta. Si la temperatura es demasiado alta, el catalizador puede desactivarse en poco tiempo, y la producción de óxidos de carbono probablemente será excesiva). Un ingeniero químico que diseñe una planta necesitará saber cuánto calor se produce para que pueda proporcionar el equipo de enfriamiento adecuado.
Debido a que no sabemos cómo llevar a cabo esta reacción, no podemos medir su cambio de entalpía directamente. Sin embargo, si tenemos las entalpías de formación para metano y metanol, podemos calcular este cambio de entalpía:
\[\ce{C(s) + 2H_2 (g) + 1/2O2 (g) -> CH3OH (g) })\nonumber \]
\[\Delta H=\Delta_fH^o\left(CH_3OH,\ g\right)\nonumber \]
\[CH_4\left(g\right)\to C\left(\mathrm{s}\right)+2\ H_2\left(g\right)\nonumber \]
\[\Delta H={-\Delta }_fH^o\left(CH_4,g\right)\nonumber \]
\[1/2 O_2\left(g\right)\to 1/2 O_2\left(g\right)\nonumber \]
\[\Delta H=-1/2 \Delta_fH^o\left(O_2,g\right)=0\nonumber \]
Sumando las reacciones da
\[\ce{ CH4 (g) + 1/2O2 (g) \to CH3OH (g)}\nonumber \]
\[\Delta H=\Delta_rH^o\nonumber \]
y sumando los cambios de entalpía da
\[\Delta_rH^o=\Delta_fH^o (CH_3OH, g) -\Delta_f H^o (CH_4, g)- 1/2 \Delta_fH^o\left(O_2,g\right)\nonumber \]
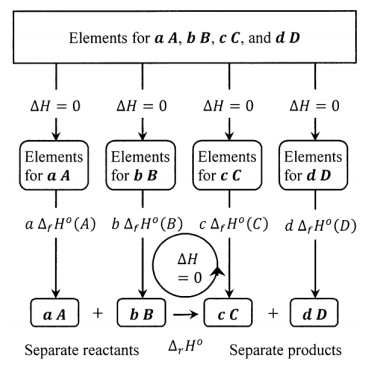
El diagrama de la Figura 2 muestra cómo estas convenciones, y el hecho de que la entalpía es una función de estado, trabajan juntas para producir, para la reacción\(aA+bB+\dots \to cC+dD+\dots\), el resultado de que la entalpía de reacción estándar viene dada por
\[\Delta_rH^o={c\ \Delta }_fH^o\left(C\right)+{d \Delta }_fH^o\left(D\right)+\dots -{a \Delta }_fH^o\left(A\right)-{b\ \Delta }_fH^o\left(B\right)-\dots\nonumber \]
Este ciclo resalta otro aspecto de las convenciones que hemos desarrollado. Nótese que\(\Delta_rH^o\) es la diferencia entre las entalpías de formación de los productos separados y las entalpías de formación de los reactivos separados. A menudo se habla de\(\Delta_rH^o\) como si fuera el cambio de entalpía que se produciría si\(a\) mezcláramos moles de\(A\) con\(b\) moles de\(B\) y la reacción procediera cuantitativamente para producir una mezcla que contenía\(c\) moles de\(C\) y\(d\) moles de\(D\). Esto suele ser una buena aproximación. Sin embargo, para relacionar rigurosamente la entalpía estándar de reacción con el cambio de entalpía que ocurriría en un sistema real en el que se produjo esta reacción, es necesario reconocer que puede haber cambios de entalpía asociados con los cambios de presión-volumen y con los procesos de mezclar los reactivos y separar los productos.
Supongamos que los reactivos y productos son gases en sus hipotéticos estados ideal-gas a 1 bar, y que llevamos a cabo la reacción mezclando los reactivos en un recipiente a presión sellado. Suponemos que entonces se inicia la reacción y que los productos se forman rápidamente, alcanzando alguna nueva presión y una temperatura elevada. (Para ser específicos, podríamos imaginar que la reacción sea la combustión del metano. Mezclaríamos cantidades conocidas de metano y oxígeno en un recipiente a presión e iniciaríamos la reacción usando una chispa eléctrica). Permitimos que la temperatura vuelva a la temperatura original de los reactivos; hay un cambio de presión acompañante.
Experimentalmente, medimos el calor evolucionado a medida que los reactivos mixtos se convierten en los productos mezclados, a la temperatura original. Para completar el proceso correspondiente al cambio de entalpía estándar, sin embargo, también debemos separar los productos y llevarlos a una presión de 1 bar. Es decir, la entalpía estándar de reacción y el cambio de entalpía que mediríamos están relacionados por la siguiente secuencia de cambios, donde la ecuación media corresponde al proceso cuyo cambio de entalpía realmente medimos.
\[{\left(aA+bB\right)}_{\mathrm{separate\ reactants\ at\ }P = 1 \text{ bar}}\to {\left(aA+bB\right)}_{\mathrm{homogenous\ mixture\ at\ }P}\nonumber \]\[\Delta H_{\mathrm{compression}}\nonumber \]
\[{\left(aA+bB\right)}_{\mathrm{separate\ reactants\ at\ }P}\to {\left(aA+bB\right)}_{\mathrm{homogeneous\ mixture\ at\ }P}\nonumber \]\[\Delta H_{\mathrm{mixing}}\nonumber \]\[{\left(aA+bB\right)}_{\mathrm{homogeneous\ mixture\ at\ }P}\to {\left(cC+dD\right)}_{\mathrm{homogeneous\ mixture\ at\ }P^*}\nonumber \]\[\Delta H_{\mathrm{measured}}\nonumber \]\[{\left(cC+dD\right)}_{\mathrm{homogeneous\ mixture\ at\ }P^*}\to {\left(cC+dD\right)}_{\mathrm{separate\ products\ at\ }P^*}\nonumber \]\[\Delta H_{\mathrm{separation}}\nonumber \]\[{\left(cC+dD\right)}_{\mathrm{separate\ products\ at\ }P^*}\to {\left(cC+dD\right)}_{\mathrm{separate\ products\ at\ }P=1\mathrm{\ bar\ }}\nonumber \]\[\Delta H_{\mathrm{expansion}}\nonumber \]
Sumando las ecuaciones de reacción da
\[{\left(aA+bB\right)}_{\mathrm{separate\ reactants\ at\ }P=1\ \mathrm{bar}}\to {\left(cC+dD\right)}_{\mathrm{separate\ products\ at\ }P=1\mathrm{\ bar\ }}\nonumber \]\[\Delta_rH^o\nonumber \]
y sumando los cambios de entalpía para la serie de etapas da el cambio de entalpía estándar para la reacción:
\[\Delta_rH^o=\Delta H_{\mathrm{compression}}+\Delta H_{\mathrm{mixing}}+\Delta H_{\mathrm{measured}} + \Delta H_{\mathrm{separation}}+\Delta H_{\mathrm{expansion}}\nonumber \]
Resulta que los cambios de entalpía para los procesos de compresión, mezcla, separación y expansión suelen ser pequeños en comparación con\(\Delta_rH^o\). Esta es la justificación principal de nuestro frecuente incumplimiento de considerarlos explícitamente. Para los gases ideales, estos cambios de entalpía son idénticamente cero. (En el Capítulo 13, vemos que los cambios de entropía para los procesos de mezcla y separación son importantes).
Cuando llamamos\(\Delta_rH^o\) al cambio de entalpía estándar “por la reacción”, nos estamos entregando un grado de licencia poética. Dado que\(\Delta_rH^o\) es una diferencia calculada entre las entalpías de los productos puros y las de los reactivos puros, la correspondiente “reacción” es un cambio puramente formal, que es una cosa claramente diferente del proceso del mundo real que realmente ocurre.