
6.1: Introducción
- Page ID
- 69407
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Con nuestro conocimiento de la estructura electrónica de los átomos estamos ahora en condiciones de entender la existencia de moléculas. Claramente, la fuerza que une los átomos entre sí para formar una molécula será, como en el caso atómico, la fuerza electrostática de atracción entre los núcleos y los electrones. En una molécula, sin embargo, nos encontramos con una fuerza de repulsión entre los núcleos además de la que existe entre los electrones. Para dar cuenta de la existencia de moléculas debemos dar cuenta del predominio de las interacciones atractivas. Daremos argumentos generales para demostrar que esto es así, primero en términos de la energía de una molécula, relativa a las energías de los átomos constituyentes, y segundo, en términos de las fuerzas que actúan sobre los núcleos en una molécula.
Para determinar qué interacciones atractivas y repulsivas son posibles en una molécula, considere una configuración instantánea de los núcleos y electrones en una molécula de hidrógeno (Figura\(\PageIndex{1}\)). Cuando los dos átomos están inicialmente muy separados (la distancia R es muy grande) las únicas interacciones potenciales son la atracción del núcleo A para el número de electrones (1) y la atracción del núcleo B para el número de electrones (2). Cuando R es comparable al diámetro de un átomo (A y B están lo suficientemente cerca como para formar una molécula) aparecen nuevas interacciones. El núcleo A atraerá ahora el electrón (2) así como (1) y de manera similar el núcleo B atraerá tanto al electrón (1) como a (2). Estas interacciones están indicadas por las cuatro líneas continuas de la Figura que\(\PageIndex{1}\) conectan pares de partículas que se atraen entre sí.
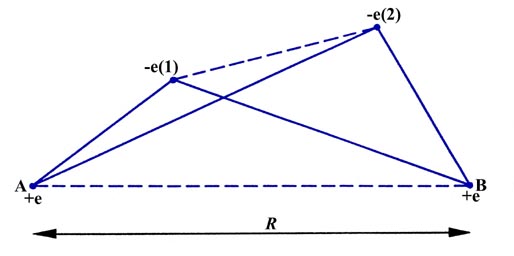
Figura\(\PageIndex{1}\): Un posible conjunto de las posiciones relativas instantáneas de los electrones y núcleos en una\(H_2\) molécula. Las líneas discontinuas representan las interacciones repulsivas entre cargas similares y las líneas continuas indican las interacciones atractivas entre cargas opuestas.
El número de interacciones atractivas se ha duplicado de lo que era cuando los átomos estaban muy separados. Sin embargo, la reducción en R introduce también dos interacciones repulsivas, indicadas por las líneas discontinuas que unen cargas de signo similar en la Figura\(\PageIndex{1}\). Los dos electrones ahora se repelen entre sí al igual que los dos núcleos. Si los dos átomos van a permanecer juntos para formar una molécula, las interacciones atractivas deben superar las repulsivas. Es claro a partir de la Figura\(\PageIndex{1}\) que las nuevas interacciones atractivas, el núcleo A que atrae electrones (2) y el núcleo B que atrae electrones (1), serán grandes solo si existe una alta probabilidad de que ambos electrones se encuentren en la región entre los núcleos. Cuando están en esta región, ambos electrones son fuertemente atraídos por ambos núcleos, en lugar de por un solo núcleo como es el caso cuando los átomos están muy separados.
Cuando la energía potencial promedio es calculada por la mecánica cuántica, se encuentra que las interacciones atractivas predominan sobre las repulsivas porque la mecánica cuántica efectivamente predice una alta probabilidad de que cada electrón esté en la región entre los núcleos. Esta consideración general de la energía demuestra que la densidad electrónica debe concentrarse entre los núcleos si se va a formar una molécula estable, pues sólo de esta manera se puede maximizar la interacción atractiva. Podemos ser mucho más específicos en nuestro análisis de este problema si discutimos una molécula desde el punto de vista de las fuerzas que actúan sobre los núcleos. Sin embargo, primero debemos exponer algunas conclusiones generales de la mecánica cuántica con respecto a los sistemas moleculares.
En el caso atómico podríamos fijar la posición del núcleo en el espacio y considerar únicamente el movimiento de los electrones con relación al núcleo. En las moléculas, los núcleos también pueden cambiar de posición entre sí. Esta complicación puede, sin embargo, ser eliminada. Los núcleos son muy masivos en comparación con los electrones y sus velocidades promedio son consecuentemente mucho más pequeñas que las que poseen los electrones. En una imagen clásica de la molécula veríamos un movimiento lento y voluminoso de los núcleos acompañado de un movimiento muy rápido de los electrones. La implicación física de esta gran disparidad en los dos conjuntos de velocidades es que los electrones pueden ajustarse inmediatamente a cualquier cambio en la posición de los núcleos. Las posiciones de los núcleos determinan el campo potencial en el que se mueven los electrones. Sin embargo, a medida que los núcleos cambian sus posiciones y por lo tanto el campo potencial, los electrones pueden ajustarse inmediatamente a las nuevas posiciones. Así, el movimiento de los electrones está determinado por dónde están los núcleos pero no por qué tan rápido se mueven los núcleos. Podemos, por este hecho, discutir los movimientos de los electrones y de los núcleos por separado.
Así, el movimiento de los electrones está determinado por dónde están los núcleos pero no por qué tan rápido se mueven los núcleos.
Para una distancia dada entre los núcleos obtenemos la energía, la función de onda y la distribución de densidad electrónica de los electrones, manteniéndose los núcleos en posiciones fijas. Entonces la distancia entre los núcleos se cambia a un nuevo valor, y se realiza de nuevo el cálculo de la energía, función de onda y distribución de densidad electrónica de los electrones. Este proceso, repetido por cada distancia internuclear posible, nos permite determinar cómo cambia la energía de los electrones a medida que se cambia la distancia entre los núcleos. Más importante para nuestra discusión actual, podemos preocuparnos solo por el movimiento de los electrones y mantener los núcleos estacionarios a algún valor particular para la distancia internuclear R.
La energía de los electrones en una molécula se cuantifica, como lo es en los átomos. Cuando los núcleos se mantienen estacionarios a algún valor fijo de R, hay una serie de niveles de energía permitidos para los electrones. Sin embargo, no hay expresiones simples para los niveles de energía de una molécula en términos de un conjunto de números cuánticos como los que encontramos para el átomo de hidrógeno. En todo caso nos ocuparemos aquí sólo del primero o más bajo de los niveles de energía para una molécula. Como en el caso de los átomos, existe una función de onda que gobierna el movimiento de todos los electrones para cada uno de los niveles de energía permitidos. Cada función de onda determina nuevamente la manera en que se distribuye la carga electrónica en el espacio tridimensional.
La distribución de densidad electrónica para una molécula se ilustra mejor por medio de un mapa de contorno, del tipo introducido anteriormente en la discusión del átomo de hidrógeno. La figura\(\PageIndex{2}\) muestra un mapa de contorno de la distribución de carga para el estado más bajo o más estable de la molécula de hidrógeno. Imagínese una molécula de hidrógeno para ser cortada por la mitad por un plano que contiene el núcleo.Se determina la cantidad de carga electrónica en cada punto del espacio, y todos los puntos que tienen el mismo valor para la densidad de electrones en el plano están unidos por una línea, una línea de contorno. También se muestra un perfil del mapa de contorno a lo largo del eje internuclear. Un perfil ilustra la variación en la densidad de carga a lo largo de un solo eje.


Figura\(\PageIndex{2}\): Un mapa de contorno de la distribución de densidad electrónica (o la distribución de carga molecular) para H2 en un plano que contiene los núcleos. También se muestra un perfil de la distribución de densidad a lo largo del eje internuclear. La separación internuclear es de 1.4 au. Los valores de los contornos aumentan en magnitud desde el más externo hacia adentro hacia los núcleos. Los valores de los contornos en este y todos los diagramas sucesivos se dan en au; 1 au = e/a o 3 = 6.749 E/å 3.
Los contornos de densidad electrónica de mayor valor se encuentran en la región de cada núcleo. Así, la carga negativa se concentra en la región de los núcleos en una molécula así como en un átomo. La siguiente concentración más alta de carga negativa se encuentra en la región entre los núcleos. Es la carga negativa en esta región la que es fuertemente atraída por ambos núcleos y que da como resultado que las interacciones atractivas superen a las repulsivas en la formación de la molécula a partir de los átomos. La mayoría de los contornos de densidad envuelven ambos núcleos. Las distribuciones de densidad de los dos átomos se han fusionado en la formación de la molécula.
El mismo mapa de contorno se obtendría para cualquier plano a través de los núcleos. Por lo tanto, en el espacio tridimensional la molécula de hidrógeno parecería ser una distribución elipsoidal de carga negativa. La mayor parte de la carga electrónica se concentra a lo largo del eje internuclear y se vuelve progresivamente más difusa a grandes distancias del centro de la molécula. Recordemos que la adición de toda la carga en cada elemento de pequeño volumen del espacio equivale al número total de electrones que en el caso de la molécula de hidrógeno es dos. El volumen de espacio encerrado por el contorno exterior de la Figura\(\PageIndex{2}\) contiene más del 99% de la carga electrónica total de la molécula de hidrógeno.