
8.6: Orbitales moleculares para moléculas heteronucleares
- Page ID
- 69455
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Los orbitales moleculares que describen el movimiento de un solo electrón en una molécula que contiene dos cargas nucleares desiguales no exhibirán las propiedades de simetría g y u del caso diatómico homonuclear. Los orbitales moleculares en el caso heteronuclear se concentrarán en general más alrededor de un núcleo que del otro. Los orbitales aún pueden clasificarse como s, p, d, etc. porque el eje molecular sigue siendo un eje de simetría.
En cálculos numéricos simples, los orbitales moleculares a veces se aproximan por la suma y diferencia de orbitales atómicos simples en cada centro, su forma limitante. Se dice que el orbital molecular se aproxima matemáticamente mediante una combinación lineal de orbitales atómicos y la técnica se conoce como el método LCAO-MO. Debe entenderse que el método LCAO-MO utilizando un número limitado de orbitales atómicos proporciona solo una aproximación al orbital molecular verdadero. El concepto de orbital molecular es completamente independiente del concepto adicional de aproximarlo en términos de orbitales atómicos, salvo en el caso de los átomos separados. Sin embargo, mediante el uso de un gran número de orbitales atómicos centrados en cada núcleo en la construcción de un solo orbital molecular se puede lograr una flexibilidad matemática suficiente para aproximarse muy de cerca a la forma exacta del orbital molecular.
Si bien la aproximación LCAO utilizando un número limitado de orbitales atómicos es en general pobre para fines cuantitativos, sí proporciona una guía útil para la predicción de las características cualitativas del orbital molecular. Hay dos condiciones simples que deben cumplirse para que los orbitales atómicos en diferentes centros interactúen significativamente y formen un orbital molecular que se deslocaliza sobre toda la molécula. Ambos orbitales atómicos deben tener aproximadamente la misma energía orbital y deben poseer las mismas características de simetría con respecto al eje internuclear. Consideraremos los orbitales moleculares en LiH, CH y HF para ilustrar cómo la teoría orbital molecular describe la unión en moléculas heteronucleares, y para ver qué tan bien se pueden racionalizar las formas de los orbitales en estas moléculas en términos de los criterios de simetría y energía expuestos anteriormente.
Los orbitales atómicos de 1 s y 2 s y el orbital 2 p que se dirige a lo largo del eje de enlace se dejan sin cambios por una rotación alrededor del eje de simetría. Por lo tanto, pueden formar orbitales moleculares de simetría en las moléculas de hidruro diatómico. Los orbitales 2 p que son perpendiculares al eje del enlace serán de simetría p y pueden formar orbitales moleculares con esta misma simetría. Las energías y propiedades de simetría de los orbitales atómicos relevantes y las configuraciones electrónicas de los átomos y moléculas se dan en la Tabla 8-2.
Cuadro 8-2.
Energía orbital atómica y propiedades de simetría
|
|
|
||||
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
| Configuraciones atómicas | Configuraciones Moleculares | ||||
| Li | 1s 2 2s 1 | LiH | 1 s 2 2 s 2 | ||
| C | 1s 2 2s 2 2p 2 | CH | 1 s 2 2 s 2 3 s 2 1 p 1 | ||
| F | 1s 2 2s 2 2p 5 | HF | 1 s 2 2 s 2 3 s 2 1 p 4 | ||
Los diagramas de densidad de los orbitales moleculares para las moléculas de LiH, CH y HF se ilustran en la Fig. 8-9.
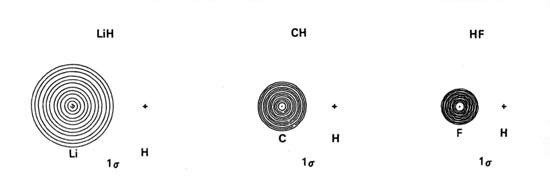
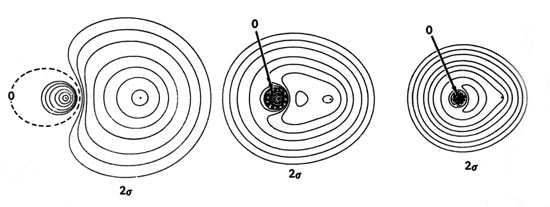
| Fig. 8-9. Mapas de contorno de las densidades de carga orbitales moleculares de los hidruros diatómicos LiH, CH, HF. Los nodos se indican mediante líneas discontinuas. Haga clic aquí para ver los valores de Countour. |
|
Las energías orbitales de 1 s de Li, C y F se encuentran muy por debajo de la orbital H 1 s. Las densidades de carga de estos orbitales de conchas internas están estrechamente ligadas a sus respectivos núcleos. No deben, por lo tanto, verse muy afectados por el campo del protón ni interactuar significativamente con la órbita H 1 s. El orbital molecular de menor energía en estas moléculas, el orbital molecular l s, debe ser esencialmente no vinculante y parecerse al orbital atómico de 1 s doblemente ocupado en Li, C y F respectivamente. Estas predicciones son corroboradas por las distribuciones de densidad orbital l s (Fig. 8-9). Consisten en contornos casi esféricos centrados en los núcleos Li, C y F, siendo el radio del contorno exterior menor que la longitud de unión en cada caso. Las fuerzas ejercidas sobre el protón por las distribuciones de carga de l s equivalen a colocar dos cargas negativas en la posición del núcleo pesado en cada caso. La densidad de carga en el orbital molecular l s simplemente criba dos de las cargas nucleares en el núcleo pesado desde el protón. Este mismo efecto de cribado se obtiene para la distribución de carga de 1 s 2 cuando las moléculas se disocian en átomos. Así, los orbitales atómicos de 1 s de Li, C y F no se ven muy afectados por la formación de la molécula y la densidad de carga de l s es no vinculante en lo que respecta al protón. Las distribuciones atómicas de l s están ligeramente polarizadas. En LiH la densidad l s se polariza alejándose del protón en un grado significativo mientras que en CH y HF se polariza ligeramente hacia el protón. Así, la densidad de carga de 1 s ejerce una fuerza antiunión sobre el núcleo de Li y una pequeña fuerza de unión sobre los núcleos C y F.
Las energías de los orbitales atómicos de 2 s disminuyen (el electrón está más estrechamente unido) de Li a F como se esperaba sobre la base del aumento de la carga nuclear efectiva de Li a F. El orbital de 2 s en Li es grande y difuso y se superpondrá extensamente con el 1 s orbital en H. Sin embargo, el electrón de 2 s en Li está considerablemente menos unido que el electrón de 1 s en H. Así, la densidad de carga del orbital molecular de 2 s en LiH se localizará en la región del protón correspondiente a la transferencia de los 2 s electrón en Li a la región de menor energía potencial ofrecida por el orbital de 1 s en H. Esto es aproximadamente correcto como lo muestra la concentración casi completa de la densidad de carga en la región del protón en el mapa de densidad orbital de 2 s para LiH. La pequeña cantidad de densidad que permanece alrededor del núcleo de Li se polariza lejos del protón. Las densidades de 1 s y 2 s están polarizadas en una dirección contraria a la dirección de transferencia de carga como se requiere en la unión iónica. La acumulación polarizada interiormente de 2 s de densidad de carga centrada en el protón se une a ambos núcleos.
El orbital molecular de 1 s en LiH es, a una buena aproximación, un orbital polarizado de 1 s doblemente ocupado en Li, y el orbital molecular de 2 s es, a una aproximación algo más pobre, un orbital de 1 s doblemente ocupado y polarizado en H. Nuestra discusión previa de la unión en LiH indicó que la unión es iónica, correspondiente a la descripción Li + (1 s 2) H - (1 s 2). La descripción orbital molecular de un enlace iónico es similar en que los orbitales moleculares en el extremo iónico se localizan en las regiones de los núcleos individuales, en lugar de ser deslocalizados sobre ambos núcleos como lo son para un enlace covalente.
La correspondencia de la energía orbital de 2 s con la energía orbital H 1 s es más cercana en el caso de C que para Li. Correspondientemente, la densidad de carga de 2 s en CH se deslocaliza sobre ambos núcleos en lugar de concentrarse en la región de un solo núcleo como está en la molécula de LiH. Hay una acumulación considerable de densidad de carga en la región de unión que es compartida por ambos núcleos. La densidad de carga de 2 s ejerce una gran fuerza de unión sobre los núcleos H y C. Esta es la descripción orbital molecular de una interacción
que es esencialmente de carácter covalente.
La energía orbital de 2 s de F es considerablemente menor que la del orbital H 1 s. La densidad de carga orbital de 2 s en HF, por lo tanto, se asemeja aproximadamente a una orbital localizada de 2 s en F. Está fuertemente polarizada y distorsionada por el protón, pero la cantidad de carga transferida a la región entre los núcleos no es tan grande como en CH. El orbital de 2 s en HF juega un papel menos importante en la unión del protón que en CH.
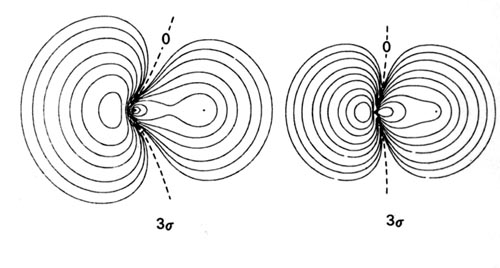
El orbital molecular de 3 s en CH y HF resultará principalmente del solapamiento del orbital 2 p s en C y F, con el orbital de 1 s en H. El carácter similar a 2 p del orbital molecular de 3 s tanto en CH como en HF es evidente en la densidad diagramas (Fig. 8-9). En CH el orbital de 1 s de H interactúa fuertemente con los orbitales de 2 s y 2 p s en C. En términos de las fuerzas ejercidas sobre los núcleos, la densidad de carga de 2 s es fuertemente vinculante tanto para C como para H, mientras que la densidad de carga de 3 s es solo muy débilmente enlazante para H y en realidad es antivinculante para el C. Este efecto antiunión es el resultado de la gran acumulación de densidad de carga en la región detrás del núcleo C.
En HF, el orbital H 1 s interactúa solo ligeramente con el orbital de 2 s en F, pero interactúa muy fuertemente con el orbital de 2 p s en la formación del orbital molecular de 3 s. La densidad de carga de 3 s ejerce una gran fuerza de unión sobre el protón. Así, el protón está unido principalmente por la densidad de carga de 2 s en CH y por la densidad de carga de 3 s en HF. La densidad de carga de 3 s en HF se centra principalmente en el núcleo F y se asemeja aproximadamente a una órbita de 2 p s. Aunque en realidad no hay contornos de densidad centrados en el protón, el protón está bien incrustado dentro de la distribución de densidad orbital. Esta es una descripción orbital molecular de un enlace altamente polar.
La densidad de carga orbital de 3 s ejerce una fuerza sobre los núcleos F y C en una dirección alejada del protón. Los orbitales moleculares que involucran orbitales p s están característicamente fuertemente polarizados en una dirección alejada del enlace en la región del núcleo en la que se centra el orbital p. Comparar, por ejemplo, los orbitales de 3 s de CH y HF con el orbital molecular de 3 s g de las moléculas diatómicas homonucleares.
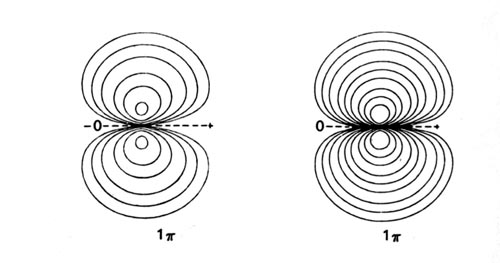
Cuando los átomos C y H están ampliamente separados, podemos considerar que el átomo de carbono tiene un electrón 2 p en el orbital de 2 p s que se encuentra a lo largo del eje del enlace, y el segundo electrón 2 p en uno de los orbitales 2 p p que son perpendiculares al vínculo. El átomo F tiene cinco electrones de 2 p y de estos uno puede colocarse en el orbital de 2 p s; los cuatro electrones restantes de 2 p ocuparán entonces completamente los orbitales de 2 p p. Los orbitales de 2 p s solo ocupados en F y C eventualmente interactúan con el orbital de 1 s solo ocupado en H para formar el orbital molecular de 3 s doblemente ocupado en HF y CH. Los 2 electrones p restantes, los de simetría p, ocuparán el orbital molecular 1 p. El átomo H no posee un orbital de simetría p en su caparazón de valencia y el orbital vacante 2 p p en H es demasiado alto en energía (-0.125 au) para interactuar significativamente con los orbitales 2 p p en C y F. Así, el 1 p molecular orbital es de tipo atómico, centrado en los núcleos F y C y es esencialmente no vinculante (Fig. 8-9). El orbital molecular 1 p se asemeja a un orbital atómico de 2 p p en cada caso, pero uno que está polarizado en la dirección del protón.
Los orbitales de 1 p de CH y HF ilustran un resultado interesante y general. En la formación de un enlace entre diferentes átomos, la densidad de carga en los orbitales s se transfiere del átomo menor al más electronegativo. Sin embargo, la densidad de carga de simetría p, si existe alguna, se transfiere invariablemente, o al menos polarizada, en dirección opuesta, hacia el átomo menos electronegativo. Aunque la cantidad de densidad de carga transferida es menor en la formación de los orbitales p que en los orbitales s, un efecto aumenta con el otro. Así, la polarización es más pronunciada en HF que en CH.
Los tres ejemplos considerados anteriormente demuestran los puntos esenciales de una descripción orbital molecular de la gama completa de enlaces químicos. En el extremo iónico de LiH, la densidad de carga del orbital molecular de enlace se localiza alrededor del protón. En CH la densidad de carga de valencia es compartida de manera más uniforme por ambos núcleos y el enlace es covalente. Los movimientos de los electrones en HF están gobernados en gran medida por el campo potencial del núcleo F. Esto se evidencia por la aparición de las distribuciones de carga orbitales moleculares. El protón está, sin embargo, abarcado por la densidad de carga de valencia y el resultado es un enlace polar.