
9.1: La aproximación Nacido-Oppenheimer simplifica la ecuación de Schrödinger para moléculas
- Page ID
- 79851
- Comprender la necesidad de introducir una aproximación como la aproximación Born-Oppenheimer para resolver sistemas multi-electrón
- Comprender las bases de la parametrización involucrada en el uso de la aproximación Born-Oppenheimer
El uso de la mecánica cuántica para predecir los patrones de unión química, las geometrías óptimas y las propiedades físicas y químicas de las moléculas es un campo de investigación amplio y activo conocido como mecánica cuántica molecular o más comúnmente como química cuántica. La teoría funcional de la densidad a la que se hace referencia en la conferencia anterior, por la que se otorgó el premio Nobel de Química en 1998, ha tenido un tremendo impacto en la química cuántica, habiendo adquirido algunas de las ponencias de esta asignatura unas 10 mil citas cada una desde su publicación. De hecho, el premio Nobel de Química de 1998 fue compartido entre Walter Kohn, uno de los inventores de la teoría funcional de la densidad y John Pople, el desarrollador de un paquete de software de química cuántica de uso común.
Los cálculos de química cuántica permiten calcular las geometrías de las moléculas así como una amplia gama de propiedades. La química cuántica también se puede utilizar de una manera novedosa, en la que los electrones son tratados mediante mecánica cuántica pero los núcleos son tratados como partículas clásicas. Utilizamos la mecánica cuántica para calcular las fuerzas internucleares, pero luego usamos estas fuerzas en la Segunda Ley de Newton para estudiar el movimiento de los núcleos durante las reacciones químicas. Esto nos da una ventana microscópica a los movimientos específicos, la danza compleja, ejecutada por los núcleos durante un proceso químico simple o complejo.
Los métodos de la química cuántica se han vuelto muy sofisticados, y existen diversos paquetes de software que se pueden descargar para realizar los cálculos de la química cuántica. Cabe señalar que estos paquetes utilizan una serie de aproximaciones para resolver la ecuación de Schrödinger porque para todas menos la más simple de las moléculas, las soluciones exactas no están disponibles. Discutiremos algunos de estos métodos, pero primero necesitamos introducir algo de la teoría subyacente.
La aproximación Nacido-Oppenheimer
La aproximación Born-Oppenheimer es uno de los conceptos básicos que subyacen a la descripción de los estados cuánticos de las moléculas. Esta aproximación permite separar el movimiento de los núcleos y el movimiento de los electrones. Esta no es una idea nueva para nosotros. Ya hicimos uso de esta aproximación en el modelo particle-in-a-box cuando explicamos los espectros de absorción electrónica de los tintes de cianina sin considerar el movimiento de los núcleos. Luego discutimos el movimiento traslacional, rotacional y vibracional de los núcleos sin incluir el movimiento de los electrones. En este capítulo examinaremos más de cerca la significación y consecuencias de esta importante aproximación. Nota, en esta discusión nuclear se refiere a los núcleos atómicos como partes de moléculas no a la estructura interna del núcleo.
La aproximación Born-Oppenheimer descuida el movimiento de los núcleos atómicos al describir los electrones en una molécula. La base física para la aproximación Born-Oppenheimer es el hecho de que la masa de un núcleo atómico en una molécula es mucho mayor que la masa de un electrón (más de 1000 veces). Debido a esta diferencia, los núcleos se mueven mucho más lentamente que los electrones. Además, debido a sus cargas opuestas, existe una fuerza mutuamente atractiva de\(Ze^2/r^2\) actuar sobre un núcleo atómico y un electrón. Esta fuerza hace que ambas partículas sean aceleradas. Dado que la magnitud de la aceleración es inversamente proporcional a la masa, a = F/m, la aceleración de los electrones es grande y la aceleración de los núcleos atómicos es pequeña; la diferencia es un factor de más de 2000. En consecuencia, los electrones se mueven y responden a las fuerzas muy rápidamente, y los núcleos no lo son. Te puedes imaginar correr una carrera de 100 yardas contra alguien cuya aceleración es 2000 veces mayor que la tuya. Esa persona literalmente podría correr círculos a tu alrededor.
Si dos partículas interactúan de alguna manera, y una es mucho más pesada que la otra, la partícula ligera se moverá esencialmente como una “esclava” de la partícula pesada. Es decir, simplemente seguirá a la partícula pesada donde quiera que vaya, y, se moverá rápidamente en respuesta al movimiento de las partículas pesadas. Como ilustración de este fenómeno, considere el simple sistema mecánico que se muestra a continuación:
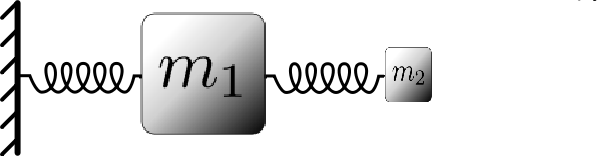
Considerando esto como un sistema clásico, esperamos que el movimiento esté dominado por la partícula grande y pesada (\(m_1\)), which is attached to a fixed wall by a spring. The small, light particle (\(m_2\), which is attached to the heavy particle by a spring will simply follow the heavy particle and execute rapid oscillations around it.
Entonces una buena aproximación es describir los estados electrónicos de una molécula pensando que los núcleos no se mueven, es decir, que son estacionarios. Los núcleos, sin embargo, pueden estar estacionarios en diferentes posiciones, por lo que la función de onda electrónica puede depender de las posiciones de los núcleos a pesar de que se descuida su movimiento.
Ahora miramos las matemáticas para ver qué se hace al resolver la ecuación de Schrödinger después de hacer la aproximación Born-Oppenheimer. Para una molécula diatómica como ejemplo, el operador hamiltoniano se agrupa en tres términos
\[\hat {H} (r, R) = \hat {T}_{nuc} (R) + \dfrac {e^2}{4\pi \epsilon _0} \dfrac {Z_A Z_B}{R} + \hat {H} _{elec} (r,R) \label {9.1.1} \]
donde
\[\hat{T}_{nuc} (R) = -\dfrac {\hbar^2}{2m_A} \nabla ^2_A - \dfrac {\hbar ^2}{2m_B} \nabla ^2_B \label {9.1.2} \]
y
\[\hat {H} _{elec} (\vec{r}, \vec{R}) = \dfrac {- \hbar ^2}{2m} \sum \limits _i \nabla ^2_i + \dfrac {e^2}{4 \pi \epsilon _0} \left ( -\sum \limits _i \dfrac {Z_A}{r_{Ai}} - \sum \limits _i \dfrac {Z_B}{r_{Bi}} + \dfrac {1}{2} \sum \limits _i \sum \limits _{j \ne i} \dfrac {1}{r_{ij}}\right ) \label{9.1.3} \]
En la Ecuación\ ref {9.1.1}, el primer término representa la energía cinética de los núcleos, el segundo término representa la repulsión de Coulomb de los dos núcleos, y el tercer término representa la contribución a la energía de los electrones, que consiste en su energía cinética, repulsión mutua entre sí, y atracción por los núcleos. \(\vec{r}\)y\(\vec{R}\) son vectores que especifican las posiciones de todos los electrones y todos los núcleos, respectivamente.
Define todos los símbolos en Ecuaciones\ ref {9.1.1} a través de\ ref {9.1.3}.
- Contestar
-
\ begin {align*}
\ hat {H} (r, R) &=\ underbrackets {\ hat {T} _ {nuc} (R)} _\ text {Término de energía cinética para núcleos} +\ underbrackets {\ frac {e^2} {4\ pi\ epsilon_o}\ frac {Z_AZ_B} {R}} _\ text {Término de repulsión para núcleos} +\ underbrackets {\ hat {H} _ {elec} (r, R)} _\ text {Hamiltoniano para electrones}\\
\ hat {T} _ {nuc } (R) &=\ underbrackets {\ frac {\ hslash^2} {2M_a}\ nAbla^2_a} _\ text {Término de energía cinética para núcleos A} -\ underbrackets {\ frac {\ hslash^2} {2m_b}\ nAbla^2_b} _\ text {Término de energía cinética para núcleos B}
\\ hat {H} _ _ elec} (\ vec {r},\ vec {R}) &=\ underbrackets {\ frac {-\ hslash^2} {2m}\ sum_ {i}\ nabla_i^2} _\ text {Cinética Término energético para electrones} +\ frac {e^2} {4\ pi\ epsilon_o}\ left (-\ underbrackets {\ sum_ {i}\ frac {Z_A} {r_ {Ai}}} _\ text {Término de atracción entre núcleos A y electrón i} -\ underbrackets {\ sum_ {i}\ frac {Z_B} {R_ {Bi {}}} _\ text {Término de atracción entre núcleos B y electrón i} +\ underbrackets {\ frac {1} {2}\ suma_ {i}\ suma_ {j\ neq i}\ frac {1} {r_ { ij}}} _\ text {Término de repulsión entre electrones}\ derecha)
\ end {alinear*}donde,\(Z_x \) es la carga de partícula x,\(m_x\) es la masa de partícula x y\(r_{xz}\) es la distancia entre partícula x y z.
Explique por qué el factor de 1/2 aparece en el último término en la Ecuación\ ref {9.1.3}.
- Contestar
-
El término 1/2 está ahí para asegurarnos de que no contemos dos veces las energías potenciales a través de las dos sumataciones. De lo contrario, añadiríamos independientemente la energía potencial del electrón 1 con el electrón 2 y la energía potencial del electrón 2 con el electrón 1. Estos son los mismos y por lo tanto hay que eliminar uno.
La aproximación Born-Oppenheimer dice que los términos de energía cinética nuclear en el hamiltoniano completo, Ecuación\ ref {9.1.1}, se pueden descuidar en la resolución de las funciones de onda y energías electrónicas. En consecuencia, la función de onda electrónica\(\varphi _e (r,R)\) se encuentra como una solución a la ecuación electrónica de Schrödinger
\[\hat {H} _{elec} (r, R) \varphi _e (r, R) = E_e (R) \varphi _e (r, R) \label {9.1.4} \]
A pesar de que se descuiden los términos de energía cinética nuclear, la aproximación Born-Oppenheimer todavía toma en cuenta la variación en las posiciones de los núcleos para determinar la energía electrónica y la función de onda electrónica resultante depende de las posiciones nucleares,\(R\). Como resultado de la aproximación Born-Oppenheimer, la función de onda molecular se puede escribir como un producto
\[\psi _{ne} (r, R) = X_{ne} (R) \varphi _e (r, R) \label {9.1.5} \]
Esta función de onda del producto se llama la función de onda Born-Oppenheimer. La función\(X_{ne} (R)\) es la función de onda vibracional, que es una función de las coordenadas nucleares\(R\) y depende tanto de los números o estados cuánticos vibracionales como electrónicos, n y e, respectivamente. La función electrónica,\(\varphi _e (r, R) \), es una función tanto de las coordenadas nucleares como electrónicas, pero sólo depende del número cuántico electrónico o estado electrónico, e. El movimiento traslacional y rotacional no se incluye aquí. Las funciones de onda traslacional y rotacional simplemente multiplican las funciones vibracionales y electrónicas en la Ecuación\ ref {9.1.5} para dar la función de onda molecular completa cuando los movimientos traslacional y rotacional no están acoplados al movimiento vibracional y electrónico.
En la Aproximación Crudo Nacido-Oppenheimer,\(R\) se establece igual a\(R_o\), la separación de equilibrio de los núcleos, y las funciones de onda electrónicas se toman como las mismas para todas las posiciones de los núcleos (es decir, los núcleos nunca se mueven). La energía electrónica,\(E_e (R)\), en la Ecuación\ ref {9.1.4} se combina con la repulsiva energía Coulomb de los dos núcleos, para formar la función de energía potencial que controla el movimiento nuclear como se muestra en la Figura 9.1.1 .
\[ V_e (R) = E_e (R) + \dfrac {e^2}{4\pi \epsilon _0} \dfrac {Z_A Z_B}{R} \label {9.1.6} \]
En consecuencia, la ecuación de Schrödinger para el movimiento vibracional es
\[( \hat {T} _{nuc} (R) + V (R) ) X_{ne} (R) = E_{ne} X_{ne} (R) \label {9.1.7} \]
Curvas y superficies de energía potencial
Anteriormente, la energía potencial se aproximaba como un potencial armónico o potencial Morse dependiendo del desplazamiento,\(R\), de los núcleos desde sus posiciones de equilibrio.
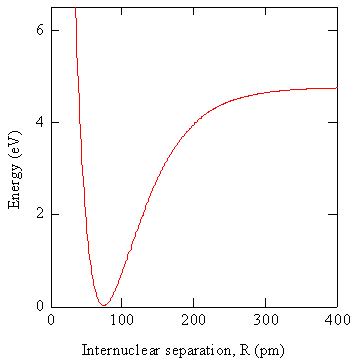
En la práctica, la ecuación electrónica de Schrödinger se resuelve utilizando aproximaciones\(R\) a valores particulares de para obtener las funciones de onda\(\varphi _e (r,R)\) y las energías potenciales\(V_e (R)\). Las energías potenciales se pueden graficar como se ilustra en la Figura 9.1.1 .
El gráfico en la Figura 9.1.1 es la energía de una molécula diatómica en función de la separación internuclear, que sirve como la función de energía potencial para los núcleos. Cuando R es muy grande hay dos átomos que interactúan débilmente. A medida que\(R\) se hace más pequeña, la interacción se vuelve más fuerte, la energía se convierte en un gran valor negativo, y decimos que se forma un enlace entre los átomos. A valores muy pequeños de\(R\), la repulsión internuclear es muy grande por lo que la energía es grande y positiva. Esta función de energía controla el movimiento de los núcleos. Anteriormente, aproximamos esta función por un potencial armónico para obtener la descripción del movimiento vibracional en términos del modelo de oscilador armónico. También podrían usarse otras formas funcionales aproximadas, por ejemplo, el potencial Morse. La posición de equilibrio de los núcleos es donde esta función es mínima, es decir, at\(R = R_0\). Si obtenemos la función de onda en\(R = R_0\) y usamos esta función para todos los valores de\(R\), hemos empleado la aproximación Crudo Born-Oppenheimer.
Relacionar la ecuación\ ref {9.1.7} con la utilizada anteriormente en nuestra descripción de las vibraciones moleculares en términos del modelo de oscilador armónico.
Mientras que la función de energía potencial\(V_e (R)\), para una molécula diatómica es una curva 1-D (Figura 9.1.1 ), las moléculas con más de dos átomos tendrán superficies de energía potencial multidimensional con dimensiones 3N-6 (o 3N-5 para molécula lineal) para el número de grados internos de libertad.
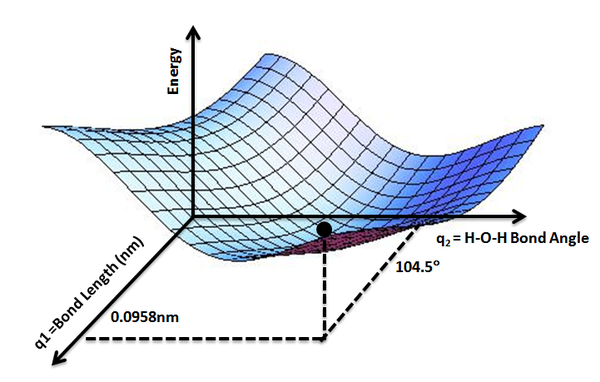
El concepto de superficie de energía potencial se puede utilizar para explorar teóricamente las propiedades de estructuras compuestas por átomos, por ejemplo, encontrar la forma de energía mínima de una molécula o calcular las velocidades de una reacción química. Cualitativamente, los diagramas de coordenadas de reacción (cortes unidimensionales a través de las superficies de energía potencial) tienen numerosas aplicaciones. Los químicos utilizan diagramas de coordenadas de reacción como ayuda analítica y pedagógica para racionalizar e ilustrar eventos cinéticos y termodinámicos. El propósito de los perfiles y superficies de energía es proporcionar una representación cualitativa de cómo varía la energía potencial con el movimiento molecular para una reacción o proceso dado.
Explicar la diferencia entre la aproximación Born-Oppenheimer y la aproximación Crudo Born-Oppenheimer.
Resumen
En esta sección comenzamos con la ecuación de Schrödinger para una molécula diatómica y la separamos en dos ecuaciones, una ecuación electrónica de Schrödinger y una ecuación nuclear de Schrödinger. Para poder hacer la separación, tuvimos que hacer una aproximación. Tuvimos que descuidar el efecto de la energía cinética nuclear sobre los electrones. El hecho de que esta suposición funcione se remonta al hecho de que las masas nucleares son mucho mayores que la masa de electrones. Luego se utilizó la solución de la ecuación electrónica de Schrödinger para proporcionar la función de energía potencial para el movimiento nuclear. La solución a la ecuación nuclear de Schrödinger proporciona las ondas vibracionales y las energías.