
15.1: Las moléculas excitadas electrónicamente pueden relajarse mediante una serie de procesos
- Page ID
- 79651
Para entender cómo funcionan los láseres primero debemos describir cómo una molécula en estado excitado puede relajarse de nuevo al estado fundamental porque la luz emitida por un láser está influenciada por estos procesos de relajación. Las dos vías de desintegración radiativa para una molécula excitada son la fluorescencia y la fosforescencia. La fluorescencia difiere de la fosforescencia en que la transición energética que es responsable de la fluorescencia no implica un cambio en la multiplicidad de espín electrónico. Así, los tiempos de vida de la fluorescencia son cortos (10 -9 - 10 -6 s). En la fosforescencia, hay un cambio en la multiplicidad de espín electrónico, lo que resulta en una vida útil más larga del estado excitado (segundo a minutos). Una descripción del espín electrónico y las diferencias entre los estados singlete y triplete aclararán la diferencia entre fluorescencia y fosforescencia.
Multiplicidad de giro: estados excitados de singlete y triplete
El principio de Exclusión Pauli establece que dos electrones en un átomo no pueden tener los mismos cuatro números cuánticos (\(n\),\(l\),\(m_l\),\(m_s\)). Por lo tanto, debido a que dos electrones pueden ocupar cada orbital, estos dos electrones deben tener estados de espín opuestos. Estos estados de giro opuestos se denominan emparejamiento de espín. Debido a este emparejamiento de espín, la mayoría de las moléculas son diamagnéticas, y no son atraídas ni repelidas por un campo magnético o eléctrico externo. Las moléculas que contienen electrones desapareados (radicales libres) tienen momentos magnéticos que son atraídos por un campo magnético o eléctrico externo.
Se dice que una molécula está en un estado singlete cuando todos los espines electrónicos están emparejados en el estado electrónico molecular, y los niveles de energía electrónica no se dividen cuando la molécula se expone a un campo magnético. Un estado de doblete ocurre cuando hay un electrón desapareado que da dos posibles orientaciones cuando se expone a un campo magnético e imparte diferentes energías al sistema. Se puede formar un estado singlete o triplete cuando un electrón se excita a un nivel de energía más alto. En un estado singlete excitado, el electrón promovido conserva la orientación de espín que tenía en el estado fundamental (es decir, emparejado). En un estado triplete excitado, el electrón promovido sufre un cambio en el espín, y así tiene la misma orientación de espín (paralela) al electrón desapareado que permanece en el estado fundamental orbital. La diferencia entre los giros de singlete de tierra, singlete excitado y triplete excitado se muestra en la Figura 15.1.1 . Los términos singlete, doblete y triplete se derivan usando la ecuación para multiplicidad, 2S+1, donde S es el momento angular de espín total (suma de todos los espines electrónicos). Los giros individuales se denotan como spin up (s = +1/2) o spin down (s = -1/2). Si tuviéramos que calcular la multiplicidad para el estado fundamental singlete o el estado excitado singlete, la ecuación sería
\[2(+1/2 + -1/2)+1 = 2(0)+1 = 1 \nonumber \]
De manera similar, la multiplicidad de giro para el estado excitado del triplete se puede calcular como
\[2(+1/2 + +1/2)+1 = 2(1)+1 =3 \nonumber \]
lo que da un estado triplete como se esperaba.

La diferencia entre una molécula en el suelo y el estado excitado del triplete es que la molécula es diamagnética en el estado fundamental y paramagnética en el estado excitado del triplete. Esta diferencia en el estado de giro hace que la transición de singlete a triplete (o triplete a singlete) sea menos probable que las transiciones de singlete a singlete. Por esta razón, la vida útil del estado triplete es más larga que la vida útil del estado singlete en un factor de aproximadamente 10 4 segundos. La transición del estado de triplete molido a excitado tiene una baja probabilidad de ocurrir, por lo que estas bandas de absorción son menos intensas que las bandas de absorción en estado singlete-singlete. Sin embargo, un estado triplete excitado puede poblarse a partir de un estado singlete excitado de ciertas moléculas. Los diagramas de Jablonski pueden ser utilizados para explicar transiciones como esta que ocurren en moléculas de fotoluminiscencia.
Diagramas de Jablonski
Aleksander Jablonski fue un académico polaco que dedicó su vida al estudio de la absorbancia molecular y emisión de luz. Desarrolló una representación gráfica que muestra las posibles consecuencias de aplicar fotones del espectro visible de la luz a una molécula particular. A estos esquemas se les conoce como diagramas de Jablonski.
Un diagrama de Jablonski es un diagrama de energía, dispuesto con energía en un eje vertical. Los niveles de energía se pueden denotar cuantitativamente, pero la mayoría de estos diagramas utilizan los niveles de energía de manera esquemática. El resto del diagrama está dispuesto en columnas. Cada columna suele representar una multiplicidad de espín específica para una especie en particular. Sin embargo, algunos diagramas dividen los niveles de energía dentro de la misma multiplicidad de espín en diferentes columnas. Dentro de cada columna, las líneas horizontales representan estados propios para esa molécula en particular. Las líneas horizontales en negrita son representaciones de los límites de los estados de energía electrónica. Dentro de cada estado de energía electrónica hay múltiples estados de energía vibratoria que pueden acoplarse con el estado electrónico. Por lo general, solo una porción de estos autoestados vibracionales están representados debido al número masivo de posibles vibraciones en una molécula. Cada uno de estos estados de energía vibracional se puede subdividir aún más en niveles de energía rotacional; sin embargo, los diagramas típicos de Jablonski omiten niveles tan intensos de detalle.
Mediante el uso de líneas rectas y onduladas, estas figuras muestran transiciones entre estados propios que ocurren a partir de la exposición de una molécula a una determinada longitud de onda de luz. Las líneas rectas muestran la conversión entre un fotón de luz y la energía de un electrón. Las líneas onduladas muestran transiciones no radiativas de electrones. Dentro de un diagrama de Jablonski, varias vías diferentes muestran cómo un electrón puede aceptar y luego disipar la energía de un fotón. Así, la mayoría de los diagramas comienzan con flechas que se originan en el estado electrónico de tierra y terminan con flechas que regresan al estado electrónico de tierra.
El diagrama de Jablonski que se dibuja a continuación es un diagrama de energía parcial que representa la energía de una molécula fotoluminiscente en sus diferentes estados energéticos. La línea horizontal más baja y más oscura representa la energía electrónica de estado fundamental de la molécula que es el estado singlete etiquetado como\(S_o\). A temperatura ambiente, la mayoría de las moléculas se encuentran en este estado. Las líneas más gruesas de la izquierda etiquetadas con S1, S2 y S3 representan los estados electrónicos excitados para la molécula en estado singlete. Las líneas más gruesas a la derecha etiquetadas con T 1, T 2 y T 3 representan estados tripletes excitados.
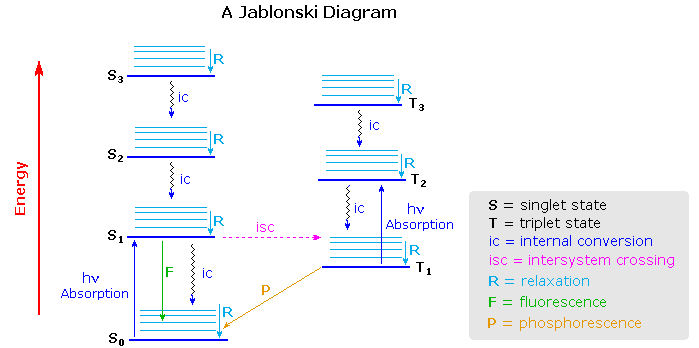
Procesos de Desactivación
Una molécula excitada puede regresar al estado fundamental mediante varias combinaciones de pasos mecánicos que se describirán a continuación y se mostrarán en la Figura 15.1.2 .El proceso de desactivación de fluorescencia y fosforescencia implica una emisión de una radiación fotónica como se muestra por la flecha recta en Figura 15.1.2 . Las flechas onduladas en la Figura 15.1.2 son procesos de desactivación sin el uso de radiación. El proceso de desactivación favorecido es la ruta que es más rápida y pasa menos tiempo en el estado excitado. Si la constante de velocidad para la relajación es más favorable en la trayectoria sin radiación, la fluorescencia será menos intensa o ausente.
Relajación y fluorescencia
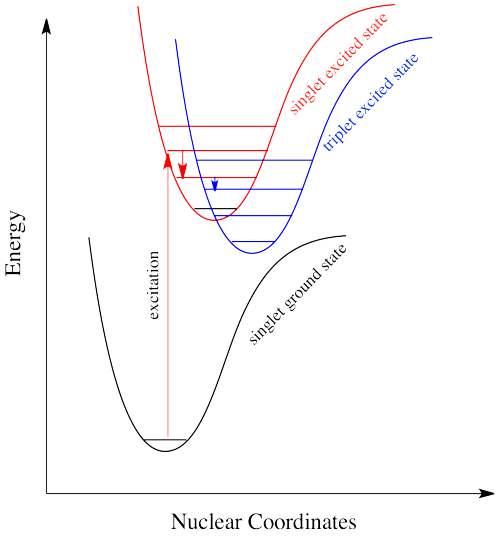
A menudo, cuando una especie en estado excitado se relaja, emitiendo un fotón, la longitud de onda del fotón es diferente a la que inicialmente condujo a la excitación. Cuando esto sucede, el fotón está invariablemente desplazado al rojo; su longitud de onda es más larga que la inicial. Esta situación se denomina “fluorescencia” (Figura\(\PageIndex{3}\)).
¿Cómo puede ser eso? ¿No se cuantifica la energía? ¿Cómo es que la molécula pierde repentinamente parte de la energía que trajo consigo el fotón original? Esta discrepancia energética está relacionada con el principio Franck-Condon de la página anterior. Cuando un electrón es promovido a un estado excitado electrónico, a menudo también termina en un estado vibracional excitado (Figura\(\PageIndex{4}\)). La energía vibratoria, sin embargo, no se intercambia exclusivamente por medio de fotones. Se puede ganar o perder a través de colisiones moleculares y transferencia de calor.
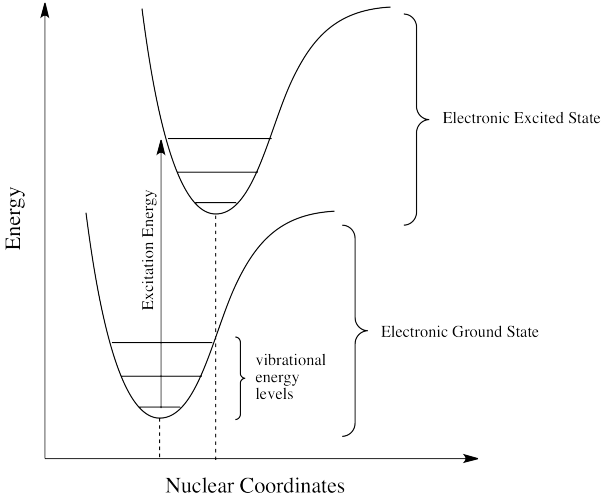
Así, una molécula de estado excitado con un electrón en un nivel vibratorio de alta energía dentro de un estado electrónico excitado podría simplemente reemitir un fotón de exactamente la misma longitud de onda que el que fue absorbido. Pero es mucho más probable que el electrón excitado se relaje al estado vibratorio más bajo dentro del estado electrónico excitado, perdiendo parte de esa energía de excitación inicial como calor. Cuando el electrón se relaja a este estado vibracional inferior, la brecha de energía entre este estado excitado y el estado fundamental es un poco menor. El fotón que se emite en la fluorescencia tendrá menor energía y longitud de onda más larga que un fotón emitido desde el estado excitado original, de mayor nivel vibracional. Ver Figura\(\PageIndex{5}\).
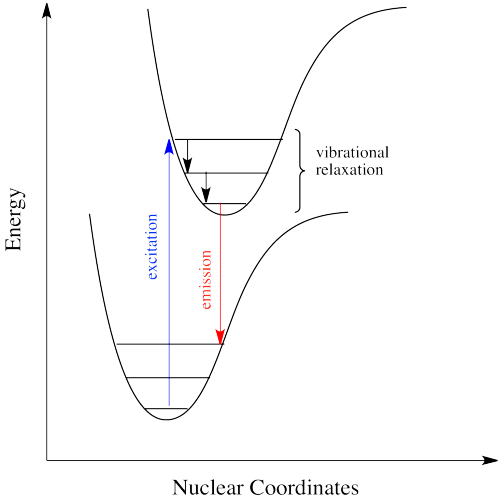
La figura\(\PageIndex{6}\) muestra las transiciones de fluorescencia de una molécula diatómica hipotética en la que la longitud del enlace de equilibrio del estado fundamental y el primer estado excitado singlete son idénticas. En esta molécula todas las absorciones implican transiciones desde el nivel vibracional más bajo del estado básico electrónico (\(v^" = 0\)) a varios niveles vibracionales en el estado electrónico excitado. Debido a que la relajación vibratoria ocurre más rápidamente que la fluorescencia, el espectro de fluorescencia está compuesto por líneas que muestran transiciones desde el nivel vibratorio más bajo del estado excitado (\(v^{'} = 0\)) a varios niveles vibracionales en el estado básico electrónico.

¿Cómo experimenta una molécula relajación vibratoria? La energía vibratoria es la energía utilizada para alargar o acortar los enlaces, o para ensanchar o apretar los ángulos de unión. Dada una molécula lo suficientemente grande, parte de esta energía vibratoria podría transferirse a longitudes de enlace y ángulos más alejados de la transición electrónica. De lo contrario, si la molécula es pequeña, puede transferir parte de su energía en colisiones con otras moléculas. En las moléculas, a medida que una molécula cae a un estado vibracional inferior, la otra saltará a un estado vibracional superior con la energía que gana. En la Figura\(\PageIndex{7}\) siguiente, la molécula roja se encuentra en un estado electrónico excitado y vibracional. En una colisión, transfiere parte de su energía vibratoria a la molécula azul.
Hay muchos ejemplos de energía que se transfiere de esta manera en la vida cotidiana. En un juego de billar, una bola de billar puede transferir su energía a otra, enviándola hacia el bolsillo. Barry Bonds puede transferir una cantidad considerable de energía a través de su bate a una pelota de béisbol, enviándola fuera del parque, así como Serena Williams puede enviar mucha energía zumbando a su hermana.
¿Cómo se compara la energía de una absorción electrónica con otros procesos? Para averiguarlo, podrías considerar la excitación de un mol entero de moléculas, en lugar de una sola molécula que absorba un solo fotón. Calcular la energía en kJ/mol para las siguientes transiciones.
- absorbancia a 180 nm (ultravioleta)
- absorbancia a 476 nm (azul)
- absorbancia a 645 nm (rojo)
- Contestar
-
Un método para llevar a cabo este cálculo:\( \dfrac{1}{x \text{nm}} \times \dfrac{1 \, \text{nm}}{1x10^{-9} \, \text{m}} \times 6.626 x 10^{-34} \text{Js} \times \dfrac{3.000 x 10^8 \text{m}}{\text{s}} \times \dfrac{6.022 x 10^{23} \text{photons}}{ 1.000 \text{mole}} \times \dfrac{1 \text{kJ}}{1000 \text{J}}\)
- 665.0 kJ/mol
- 251.5 kJ/mol
- 185.6 kJ/mol
¿Cómo se compara la energía de una excitación entre estados vibratorios con la de una excitación electrónica? Por lo general, las absorciones infrarrojas se reportan en cm -1, que es simplemente lo que parece: el recíproco de la longitud de onda en cm. Debido a que la longitud de onda y frecuencia están inversamente relacionadas, los números de onda se consideran una unidad de frecuencia. Calcular la energía en kJ/mol para las siguientes transiciones.
- absorbancia a 3105 cm -1
- absorbancia a 1695 cm -1
- absorbancia a 963 cm -1
- Contestar
-
Un método para llevar a cabo este cálculo:\( x \text{cm}^{-1} \times \dfrac{100 \, \text{cm}}{1 \, \text{m}} \times 6.626 x 10^{-34} \text{Js} \times \dfrac{3.000 x 10^8 \text{m}}{\text{s}} \times \dfrac{6.022 x 10^{23} \text{photons}}{ 1.000 \text{mole}} \times \dfrac{1 \text{kJ}}{1000 \text{J}}\)
- 37.14 kJ/mol
- 20.27 kJ/mol
- 11.52 kJ/mol
Al comparar estas respuestas con las del Ejercicio\(\PageIndex{1}\), podemos ver que la diferencia de energía entre los estados vibracionales es un orden de magnitud menor que la diferencia de energía entre los estados electrónicos.
Conversión interna
Si los electrones pueden llegar a un estado de menor energía, y emitir un poco de energía a la vez, relajándose para bajar y bajar los niveles vibracionales, ¿necesitan emitir un fotón en absoluto? Tal vez puedan relajarse todo el camino hasta el estado base a través de la relajación vibracional. Ese es sin duda el caso. Dados muchos niveles de energía vibratoria, y un estado excitado que es lo suficientemente bajo en energía para que algunos de sus niveles vibracionales más bajos se superpongan con algunos de los niveles vibracionales más altos del estado fundamental, un electrón puede relajarse de un estado electrónico excitado de nuevo al estado fundamental sin liberar un fotón ( Figura\ (\ PageIndex {8}).

A este evento se le llama una “transición sin radiación”, porque ocurre sin liberación de un fotón. El electrón simplemente se desliza desde un estado vibracional bajo del estado electrónico excitado a un estado vibracional alto del estado básico electrónico. Si el electrón simplemente sigue cayendo un nivel vibracional en un tiempo de regreso al estado fundamental, el proceso se llama “conversión interna”.
La conversión interna tiene una consecuencia importante. Debido a que la absorción de luz UV y visible puede resultar en la transferencia de energía a estados vibratorios, gran parte de la energía que se absorbe de estas fuentes se convierte en calor. Eso puede ser algo bueno si resulta que eres una iguana marina tratando de calentarse al sol después de un chapuzón en el helado Pacífico. También puede ser algo complicado si usted es un químico de procesos tratando de escalar una reacción fotoquímica para la producción comercial de un farmacéutico, porque hay que asegurarse de que el sistema tenga la refrigeración adecuada disponible.
Cruce intersistémico
El cruce intersistémico es un proceso que lleva a que el electrón quede atrapado entre el estado excitado y el estado fundamental. Así como, poco a poco, la relajación vibratoria puede llevar al electrón de nuevo a la superficie de energía del estado fundamental, también puede conducir al electrón a estados que son intermedios en energía en los que la multiplicidad de espín ha cambiado.
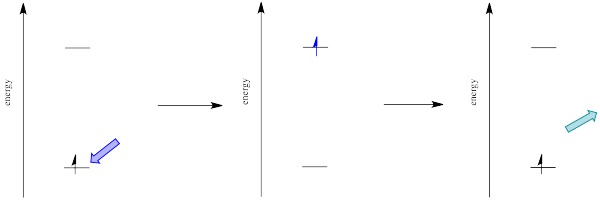
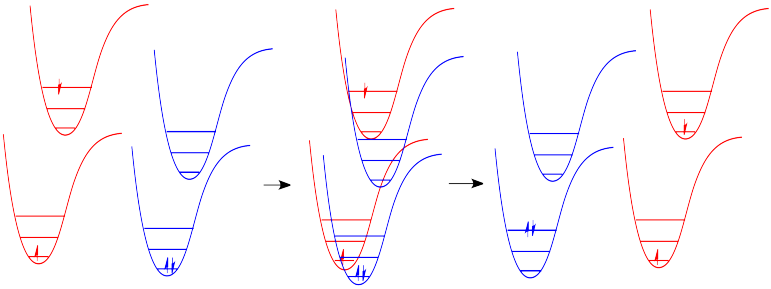
Por ejemplo, supongamos que una molécula orgánica sufre excitación electrónica. Generalmente, las moléculas orgánicas no tienen electrones desapareados. Sus estados terrestres son estados singlete. De acuerdo con una de las reglas de selección para la excitación electrónica, el estado excitado tampoco debe tener electrones desapareados. Es decir, el giro en el electrón que se excita es el mismo después de la excitación que antes de la excitación.
Sin embargo, ese no es el estado de energía más bajo posible para ese electrón. Cuando pensamos en el llenado orbital atómico, hay una regla que gobierna el giro sobre los electrones en orbitales degenerados: en el estado de energía más baja, el giro se maximiza (regla de Hund). En otras palabras, cuando dibujamos una imagen de la configuración de electrones de valencia del nitrógeno, mostramos los tres electrones p de nitrógeno cada uno en su propio orbital, con sus espines paralelos.

En la Figura\(\PageIndex{9}\), el diagrama de la izquierda, con tres electrones desapareados, todos con espines paralelos, muestra un nitrógeno en el estado de espín del cuarteto. Este es el estado más estable. Tener uno de esos giros apuntando de otra manera resultaría en un estado de giro diferente y de mayor energía. El diagrama de la derecha muestra un estado de espín diferente a diferente, en el que un par de electrones en el nivel p está emparejado por espín, uno hacia arriba y otro hacia abajo, a pesar de que están en diferentes orbitales p. Eso dejaría a un electrón sin pareja opuesta, por lo que el nitrógeno estaría en estado de giro doblete. El estado de giro a la izquierda es menor en energía que el estado de la derecha. Esa es solo una de las reglas de la mecánica cuántica (regla de Hund): maximizar el giro cuando los orbitales degenerados están ocupados individualmente.
Un argumento similar se puede hacer para una molécula con el estado triplete menor en energía que el estado singlete, como se muestra en la Figura\(\PageIndex{10}\). ¿Por qué el electrón no se excitó al estado triplete en primer lugar? Esa es una transición electrónica prohibida (improbable). Pero deslizarse hacia abajo vibracionalmente sobre el estado triplete desde el estado excitado singlete no lo es, porque no implica absorción o emisión de un fotón.
El cruce entre sistemas puede tener importantes consecuencias en la química de las reacciones, ya que permite acceder a estados tripletes que normalmente no están disponibles en muchas moléculas. Debido a que los estados tripletes presentan electrones desapareados, su reactividad suele ser tipificada por procesos radicales. Eso significa que se puede acceder a un conjunto adicional de reacciones a través de este proceso.
Fosforescencia: una transición sin radiación seguida de emisión
El cruce entre sistemas es una forma en que un sistema puede terminar en un estado de triplete excitado. A pesar de que este estado es menor en energía que un estado excitado singlete, no se puede acceder directamente a él a través de la excitación electrónica porque eso violaría la regla de selección de giro (\ Delta S=0\). Una vez que se ha producido el cruce entre sistemas, el proceso de recuperación del estado base se ralentiza drásticamente. El camino rápido de regreso al estado fundamental es emitir un fotón. Pero debido a que eso implicaría un cambio en el estado de giro, no está permitido. Hablando de manera realista, eso significa que lleva mucho tiempo. Por “mucho tiempo”, podríamos significar unos segundos, varios minutos, o posiblemente incluso horas. Eventualmente, el electrón puede volver a caer, acompañado de la emisión de un fotón. Esta situación se llama “fosforescencia” (Figura\ (\ PageIndex {11}).
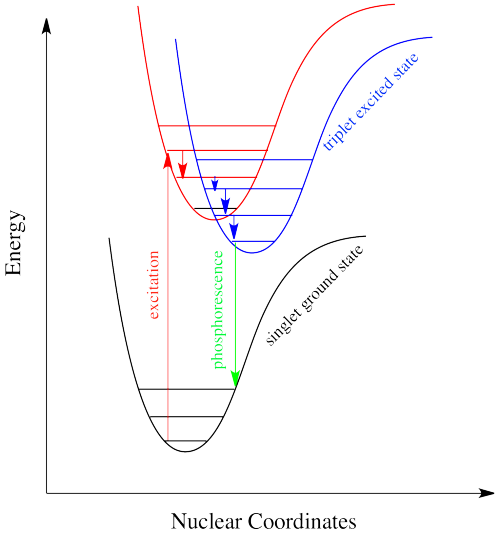
Muchas plantas y animales utilizan la fosforescencia como medio de señalización. Las moléculas que muestran fosforescencia también suelen incorporarse a los juguetes y camisas para que brillen en la oscuridad.
Conversión Externa
La desactivación del estado electrónico excitado también puede implicar la interacción y transferencia de energía entre el estado excitado y el disolvente o soluto en un proceso llamado conversión externa. La baja temperatura y la alta viscosidad conducen a una mayor fluorescencia porque reducen el número de colisiones entre moléculas, lo que ralentiza este tipo de proceso de desactivación.
Tasas de absorción y emisión
La siguiente tabla compara las tasas de absorción y emisión de fluorescencia y fosforescencia. La tasa de absorción de fotones es muy rápida. La emisión de fluorescencia se produce a una velocidad más lenta. Dado que el triplete a singlete (o inverso) es una transición prohibida, lo que significa que es menos probable que ocurra que la transición singlete a singlete, la tasa de triplete a singlete suele ser más lenta. Por lo tanto, la emisión de fosforescencia requiere más tiempo que la fluorescencia.
| Proceso | ¿Proceso Radiativo? | Transición | Escala de tiempo (seg) |
|---|---|---|---|
| Absorción de luz (excitación) | si | S 0 → S n | ca. 10 -15 (instantáneo) |
| Conversión interna | no | S n → S 1 | 10 -14 a 10 -11 |
| Relajación Vibracional | no | S n * → S n | 10 -12 a 10 -10 |
| Cruce intersistémico | no | S 1 → T 1 | 10 -11 a 10 -6 |
| Fluorescencia | si | S 1 → S 0 | 10 -9 a 10 -6 |
| Fosforescencia | si | T 1 → S 0 | 10 -3 a 100 |
| Decaimiento no radiativo a estado fundamental (conversión interna) | no | S 1 → S 0 T 1 → S 0 |
10 -7 a 10 -5 10 -3 a 100 |
Referencias
- D. A. Skoog, et al. “Principios del Análisis Instrumental” 6ª Edición, Thomson Brooks/Cole. 2007
- D. C. Harris y M.D. Bertolucci “Simetría y espectroscopia, una introducción a la espectroscopia vibracional y electrónica” Dover Publications, Inc., Nueva York. 1989.
Colaboradores y Atribuciones
Chris P Schaller, Ph.D., (College of Saint Benedict / Saint John's University)
- Diana Wong (UCD)
- Tom Neils (Grand Rapids Community College)