
2.1: Modelo de electrones libres de polienos
- Page ID
- 70944

Los problemas de tipo partícula en caja proporcionan modelos importantes para varias situaciones químicas relevantes
El modelo de partículas en caja para movimiento en una o dos dimensiones discutidas anteriormente obviamente puede extenderse a tres dimensiones. Para dos y tres dimensiones, proporciona una imagen cruda pero útil para estados electrónicos en superficies (es decir, cuando el electrón puede moverse libremente sobre la superficie pero no puede escapar al vacío o penetrar profundamente en el sólido) o en cristales metálicos, respectivamente. Digo cristales metálicos porque es en tales sistemas donde los electrones de valencia más externos son razonablemente bien tratados como moviéndose libremente en lugar de estar fuertemente unidos a un orbital de valencia en uno de los átomos constituyentes o dentro de enlaces químicos localizados a átomos vecinos.
El libre movimiento dentro de un volumen esférico como lo discutimos en el Capítulo 1 da lugar a funciones propias que también se utilizan en la física nuclear para describir los movimientos de neutrones y protones en los núcleos. En el llamado modelo de núcleos de concha, los neutrones y protones llenan orbitales separados\(s\),\(p\),\(d\), etc. (refiérase de nuevo al Capítulo 1 para recordar cómo se expresan estos orbitales en términos de funciones esféricas de Bessel y cuáles son sus energías) con cada tipo de nucleón obligado a obedecer el Principio de exclusión de Pauli (es decir, tener no más de dos nucleones en cada orbital porque los protones y neutrones son Fermiones). Por ejemplo,\(^4He\) tiene dos protones en\(1s\) orbitales y 2 neutrones en\(1s\) orbitales, mientras que\(^3He\) tiene dos\(1s\) protones y un\(1s\) neutrón. Para recordarles, muestro en la Figura 2. 1 las formas angulares que caracterizan\(s\),\(p\), y\(d\) orbitales.

Figura 2.1. Las formas angulares de\(s\)\(p\), y\(d\) funciones
Este mismo modelo de caja esférica también se ha utilizado para describir los electrones de valencia en nanogrupos cuasiesféricos de átomos metálicos como\(Cs_n\),\(Cu_n\),\(Na_n\),\(Au_n\),\(Ag_n\), y sus iones positivos y negativos. Debido a la naturaleza metálica de estas especies, sus electrones de valencia son esencialmente libres para deambular sobre todo el volumen esférico del cúmulo, lo que hace que este sencillo modelo sea bastante efectivo. En este modelo, se piensa que cada electrón de valencia es libre para vagar dentro de una esfera de radio\(R\) (es decir, tener un potencial que es uniforme dentro de la esfera e infinito fuera de la esfera).
Los orbitales que resuelven la ecuación de Schrödinger dentro de una caja esférica de este tipo no son los mismos en sus formas radiales que los orbitales\(s\)\(p\)\(d\),,, etc. de los átomos porque, en los átomos, existe un potencial radial de Coulomb atractivo adicional\(V(r) = -Ze^2/r\) presente. En el Capítulo 1, mostramos cómo las funciones radiales de partículas en una esfera pueden expresarse en términos de funciones esféricas de Bessel. Además, el patrón de niveles de energía, que se mostró en el Capítulo 1 relacionado con los valores de x a los que\(j_L(x)\) desaparecen las funciones esféricas de Bessel, no son los mismos que en los átomos, nuevamente porque los potenciales radiales difieren. Sin embargo, las formas angulares del problema de la caja esférica son las mismas que en la estructura atómica porque, en ambos casos, el potencial es independiente de\(\theta\) y\(\phi\). Como indican las gráficas orbitales mostradas anteriormente, las formas angulares de s, p y\(d\) orbitales muestran un número variable de superficies nodales. Los\(s\) orbitales no tienen ninguno,\(p\) los orbitales tienen uno, y\(d\) los orbitales tienen dos. Análogamente a cómo el número de nodos relacionados con la energía total de la partícula restringida al\(xy\) plano, el número de nodos en las funciones de onda angular indica la cantidad de energía rotacional angular u orbital. Los orbitales de\(s\) forma no tienen energía angular, los de\(p\) forma tienen menos que hacer\(d\) orbitales, etc.
Resulta que el patrón de niveles de energía derivado de este modelo de partículas en una caja esférica puede ofrecer descripciones razonablemente precisas de lo que se observa experimentalmente. En particular, cuando un cúmulo (o ion clúster) tiene una configuración electrónica de carcasa cerrada en la que, para un número cuántico radial dado\(n\), todos los\(s\)\(d\) orbitales asociados a los que\(n\) están doblemente ocupados, se observan conglomerados metálicos nanoscópicos para mostrar\(p\) estabilidad especial (e.g., falta de reactividad química, gran energía de desprendimiento de electrones). A veces se dice que los clústeres que producen tales configuraciones electrónicas de carcasa cerrada tienen tamaños de números mágicos. La expresión del nivel de energía dada en el Capítulo 1
\[E_{L,n} = V_0 + (z_{L,n})^2 \dfrac{h^2}{2mR^2} \tag{2.1}\]
para un electrón que se mueve dentro de una esfera de radio\(R\) (y que tiene un potencial relativo al vacío de\(V_0\)) se puede utilizar para modelar las energías de electrones dentro de nano-clusters metálicos. Cada electrón ocupa un orbital que tiene números cuánticos\(n\)\(L\), y\(M\), con las energías de los orbitales dadas anteriormente en términos de los ceros\(\{z_{L,n}\}\) de las funciones esféricas de Bessel. Las características espectrales de los nano-clusters se determinan entonces por la brecha de energía entre el orbital ocupado más alto y el más bajo desocupado y se pueden sintonizar cambiando el radio (\(R\)) del clúster o la carga (es decir, el número de electrones) del clúster.
Otra aplicación muy útil de los problemas del modelo tratados en el Capítulo 1 es la partícula unidimensional en una caja, que proporciona una imagen cualitativamente correcta para el movimiento de\(\pi\) electrones a lo largo de los\(p_{\pi}\) orbitales de polienos deslocalizados. La dimensión cartesiana corresponde al movimiento a lo largo de la cadena deslocalizada. En dicho modelo, la longitud de la caja\(L\) está relacionada con la longitud del enlace carbono-carbono\(R\) y el número\(N\) de centros de carbono involucrados en la red deslocalizada\(L=(N-1) R\). En la Figura 2.2, se representa una red conjugada de este tipo que involucra nueve centros. En este ejemplo, la longitud de la caja sería ocho veces la longitud del enlace C-C.

Los autoestados\(\psi_n(x)\) y sus energías\(E_n\) representan orbitales en los que se colocan los electrones. En el caso de ejemplo, si hay nueve\(\pi\) electrones presentes (por ejemplo, como en el radical 1,3,5,7-nonatetraeno), el estado electrónico básico estaría representado por una función de onda total que consiste en un producto en el que los cuatro\(\psi\) más bajos están doblemente ocupados y el quinto\(\psi\) está ocupado individualmente:
\[\Psi = \psi_1 \alpha\psi_1\beta \psi_2 \alpha \psi_2 \beta \psi_3 \alpha \psi_3\beta \psi_4 \alpha \psi_4 \beta \psi_5 \alpha. \tag{2.2}\]
Los estados de momento angular de giro\(z\) -componente de los electrones están etiquetados\(\alpha\) y\(\beta\) como se discutió anteriormente.
Escribimos la función de onda total anterior como una función de onda producto porque el hamiltoniano total involucra las energías cinéticas más potenciales de nueve electrones. En la medida en que esta energía total puede representarse como la suma de nueve energías separadas, una por cada electrón, la hamiltoniana permite una separación de variables
\[H \cong \sum_{j=1}^9 H(j) \tag{2.3}\]
en el que cada H (j) describe la energía cinética y potencial de un electrón individual. Por supuesto, el Hamiltoniano completo contiene potenciales de interacción electrón-electrón Coulomb\(e^2/r_{i,j}\) que no se pueden escribir en esta forma aditiva. Sin embargo, como trataremos en detalle en el Capítulo 6, a menudo es posible aproximar estas interacciones electrón-electrón en una forma que es aditiva.
Recordemos que cuando una ecuación diferencial parcial no tiene operadores que acoplan sus diferentes variables independientes (es decir, cuando es separable), se pueden utilizar métodos de separación de variables para descomponer sus soluciones en productos. Así, la aditividad (aproximada) de\(H\) implica que las soluciones de\(H \psi = E \psi\) son productos de soluciones para
\[H (j) \psi (\textbf{r}_j) = E_j \psi(\textbf{r}_j). \tag{2.4}\]
Los dos estados más\(\pi\pi^*\) excitados corresponderían a estados de la forma
\[\psi^* = \psi_1\alpha \psi_1\beta \psi_2\alpha \psi_2\beta \psi_3\alpha \psi_3\beta \psi_4\alpha \psi_5\beta \psi_5\alpha, \tag{2.5a}\]
y
\[\psi'^* = \psi_1\alpha \psi_1\beta \psi_2\alpha \psi_2\beta \psi_3\alpha \psi_3\beta \psi_4\alpha \psi_4\beta \psi_6\alpha,\tag{2.5b}\]
donde los orbitales espín (orbitales multiplicados por\(\alpha\) o\(\beta\)) que aparecen en los productos anteriores dependen de las coordenadas de los diversos electrones. Por ejemplo,
\[\psi_1\alpha \psi_1\beta \psi_2\alpha \psi_2\beta \psi_3\alpha \psi_3\beta \psi_4\alpha \psi_5\beta \psi_5\alpha \tag{2.6a}\]
denota
\[ \psi_1\alpha(\textbf{r}_1) \psi_1\beta (\textbf{r}_2) \psi_2\alpha (\textbf{r}_3) \psi_2\beta (\textbf{r}_4) \psi_3\alpha (\textbf{r}_5) \psi_3\beta (\textbf{r}_6) \psi_4a (\textbf{r}_7)\psi_5\beta (\textbf{r}_8) \psi_5\alpha (\textbf{r}_9). \tag{2.6b}\]
Las energías de excitación electrónica desde el estado fundamental hasta cada uno de los estados excitados anteriores dentro de este modelo serían
\[\Delta{E^*} = \dfrac{ \pi^2 \hbar^2}{2m} \left[ \dfrac{5^2}{L^2} - \dfrac{4^2}{L^2}\right] \tag{2.7a}\]
y
\[\Delta{E'^*} = \dfrac{ \pi^2 \hbar^2}{2m} \left[ \dfrac{6^2}{L^2} - \dfrac{5^2}{L^2}\right]. \tag{2.7b}\]
Resulta que este modelo simple de energías\(\pi\) -electrónicas proporciona una imagen cualitativamente correcta de tales energías de excitación. Su simplicidad permite, por ejemplo, sugerir fácilmente cómo varía el color de una molécula (como se refleja en el color complementario de la luz que absorbe la molécula) a medida que varía la longitud\(L\) de conjugación de la molécula. Es decir, las moléculas conjugadas más largas tienen orbitales de menor energía porque\(L^2\) aparece en el denominador de la expresión energética. Como resultado, las moléculas conjugadas más largas absorben luz de menor energía que las moléculas más cortas.
Este modelo simple de partículas en caja no produce energías orbitales que se relacionan con las energías de ionización a menos que se especifique el potencial dentro de la caja. Elegir el valor de este potencial\(V_0\) que existe dentro de la caja tal que\(V_0 + \dfrac{\pi^2 \hbar^2}{2m} \dfrac{5^2}{L^2}\) sea igual a menos la energía de ionización más baja del radical 1,3,5,7-nonatetraeno, da niveles de energía (as\(E = V_0 + \dfrac{\pi^2 \hbar^2}{2m} \dfrac{n^2}{L^2}\)), que luego pueden ser utilizados como aproximaciones a las energías de ionización.
Los\(\pi\) orbitales moleculares individuales
\[\psi_n = \sqrt{\dfrac{2}{L}} \sin\Big(\dfrac{n\pi x}{L}\Big) \tag{2.8}\]
se representan en la Figura 2.3 para un modelo del sistema 1,3,5\(\pi\) hexatrieno-orbital para el cual la longitud de la caja\(L\) es cinco veces la distancia\(R_{CC}\) entre pares vecinos de átomos de carbono. La magnitud del orbital atómico centrado en el\(k^{th}\) átomo de C en el\(n^{th}\)\(\pi\) orbital molecular viene dada por
\[\sqrt{\dfrac{2}{L}} \sin\Big(\dfrac{n\pi(k-1)R_{CC}}{L}\Big).\]
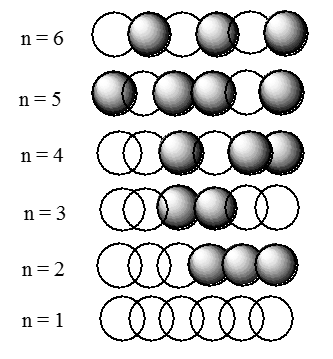
Figura 2.3. Las fases de los seis orbitales moleculares de una cadena que contiene seis átomos.
En esta figura, la amplitud positiva se denota por las esferas claras, y la amplitud negativa se muestra por las esferas oscurecidas. Cuando dos esferas de sombreado similar se superponen, la función de onda tiene mayor amplitud (es decir, hay una interacción de unión); donde dos esferas de diferente sombreado se superponen, se produce un nodo (es decir, hay interacción antiunión). Una vez más, observamos que el número de nodos aumenta a medida que uno va desde el orbital de menor energía hasta los orbitales de mayor energía. Se anima una vez más al lector a tener presente esta característica ubicua de las funciones de onda mecánica cuántica.
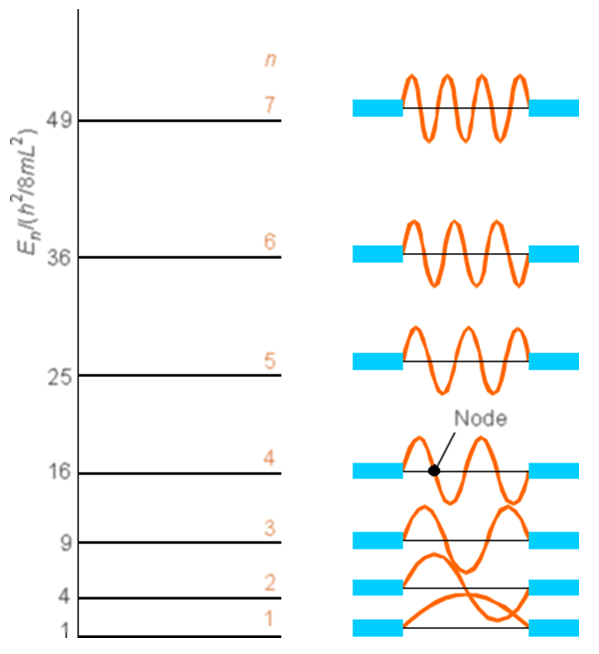
Este modelo simple permite estimar las densidades de espín en cada centro de carbono y proporciona información sobre qué centros deberían ser más susceptibles al ataque electrófilo o nucleófilo. Por ejemplo, el ataque radical al\(C_5\) carbono del sistema de nonatetraeno de nueve átomos descrito anteriormente sería más fácil para el estado fundamental\(\psi\) que para cualquiera\(\psi^*\) o\(\psi'^*\). En el primero, la densidad de espín desapareada reside en\(\psi_5\) (que varía como\(\sin(5\pi x/8R_{CC}\)) por lo que es distinta de cero en\(x = L/2\)), que tiene una amplitud distinta de cero en el\(C_5\) sitio\(x= L/2 = 4R_{CC}\). En\(\psi^*\) y\(\psi'*\), la densidad desapareada está en\(\psi_4\) y\(\psi_6\), respectivamente, ambos tienen densidad cero en\(C_5\) (porque el pecado (NPx/8Rcc) desaparece para\(n = 4\) o\(6\) en\(x = 4R_{CC}\)). Las gráficas de las funciones de onda\(n\) para variar de 1 a 7 se muestran en otro formato en la Figura 2.4 donde se enfatiza el patrón nodal.
Espero que a estas alturas el estudiante no esté tentado a preguntar cómo el electrón pasa de una región de alta amplitud, a través de un nodo, a otra región de alta amplitud. Recuerde, tales preguntas se emiten en lenguaje newtoniano clásico y no son apropiadas al abordar las propiedades onduladas de la mecánica cuántica.
Colaboradores y Atribuciones
Jack Simons (Henry Eyring Scientist and Professor of Chemistry, U. Utah) Telluride Schools on Theoretical Chemistry
Integrated by Tomoyuki Hayashi (UC Davis)