
10.1: Sondas de espín y etiquetas de nitróxido
- Page ID
- 76877
Sondas giratorias y etiquetas
Las sondas de espín son especies paramagnéticas estables que se mezclan a una muestra para obtener información estructural o dinámica sobre su entorno y, por lo tanto, indirectamente sobre la muestra. Los marcadores de espín son sondas de espín que están unidas covalentemente a una molécula de interés, a menudo en un sitio específico. En comparación con la caracterización más directa de la estructura y la dinámica por otras técnicas, la espectroscopia EPR en sondas de espín puede ser capaz de acceder a otras escalas de longitud y tiempo o puede ser aplicable en estados de agregación o entornos donde estas otras técnicas exhiben baja resolución o no producen ninguna señal. El etiquetado de espín dirigido al sitio (SDSL) tiene la ventaja de que ya se conoce la asignación de la señal a la estructura molecular primaria y que un sitio específico en un sistema complejo puede estudiarse sin perturbación de las señales de otras partes del sistema. Este enfoque se beneficia de la rareza de los centros paramagnéticos. Por ejemplo, muchas proteínas y la mayoría de los ácidos nucleicos y lípidos son diamagnéticos. Si se introduce una etiqueta giratoria en un sitio seleccionado, la información de EPR es específica de este sitio en particular.
En principio, cualquier especie paramagnética estable puede servir como sonda de espín. Algunos iones metálicos paramagnéticos pueden sustituir a los iones diamagnéticos nativos del sistema en estudio, ya que tienen carga y radio iónico similares o con propiedades de complejación similares a las de los iones nativos. Esto se aplica a\(\mathrm{Mn}(\mathrm{II})\), que a menudo puede sustituir\(\mathrm{Mg}(\mathrm{II})\) sin afectar la función de proteínas o ácidos nucleicos, o iones lantánidos Ln (III), que se unen a sitios Ca (II). Los iones metálicos paramagnéticos también pueden unirse a proteínas mediante ingeniería genética de sitios de unión con aminoácidos coordinantes, tales como histidina, o mediante la unión dirigida al sitio de un ligando metálico a la biomolécula. Dichos enfoques se utilizan para iones lantánidos, en particular Gd (III) y Cu (II).
Para muchos enfoques de sonda de espín, los radicales orgánicos son más adecuados que los iones metálicos, ya que en los radicales el electrón desapareado tiene un contacto más cercano con su entorno (los ligandos filtran el acceso ambiental de iones metálicos, en particular para iones lantánidos) y los espectros EPR son más estrechos, lo que permite algunos experimentos que no se pueden realizar en especies con espectros muy amplios. Entre los radicales orgánicos, los nitróxidos son la clase más versátil de sondas de espín, principalmente por su tamaño relativamente pequeño, comparable a un grupo lateral de aminoácidos o nucleobase, y debido a la anisotropía hiperfina y\(g\) tensor de una magnitud que es conveniente para estudiar la dinámica ( Sección 10.1.4). Los radicales triarilmetilo (TAM) son químicamente incluso más inertes que los radicales nitróxido y tienen tiempos de relajación más lentos en solución líquida. Actualmente están mucho menos en uso que los radicales nitróxido, principalmente porque no están disponibles comercialmente y son mucho más difíciles de sintetizar que los radicales nitróxido.
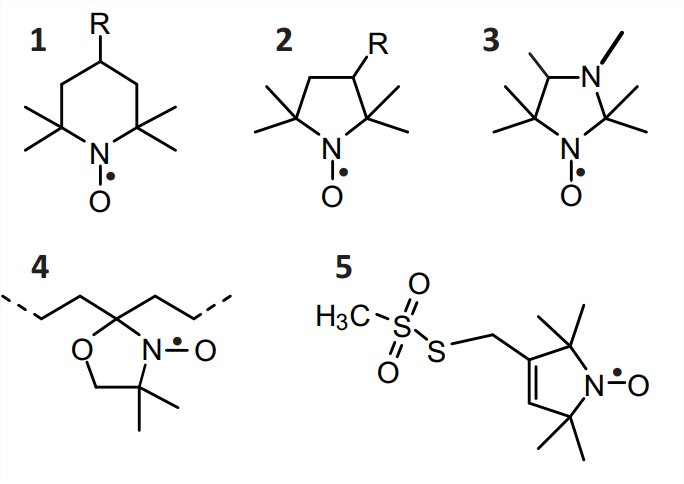
Radicales Nitróxido
El radical nitróxido se define por el\(\mathrm{N}-\mathrm{O}^{\bullet}\) grupo, que es isoelectrónico con el grupo carbonilo\((\mathrm{C}=\mathrm{O})\) y así puede ser reemplazado en cálculos aproximados de campo de fuerza y dinámica molecular por un\(\mathrm{C}=\mathrm{O}\) grupo. El electrón desapareado se distribuye sobre ambos átomos, lo que contribuye a la estabilidad radical, con una ligera preferencia por el átomo de oxígeno. Los radicales nitróxido se vuelven estables en la escala de tiempo de meses o años si ambas\(\alpha\) posiciones están protegidas estéricamente, por ejemplo, uniendo dos grupos metilo a cada uno de los\(\alpha \mathrm{C}\) átomos (Fig. 10.1). Los nitróxidos de este tipo son térmicamente estables hasta temperaturas de aproximadamente\(140^{\circ} \mathrm{C}\), pero se reducen fácilmente a las hidroxilaminas correspondientes, por ejemplo por el ácido ascórbico, y son inestables a pH muy bajo y muy alto. Los nitróxidos con anillos de cinco miembros (estructuras\(\mathbf{2}, \boldsymbol{3}\), y\(\mathbf{5}\)) tienden a ser químicamente más estables que aquellos con anillos de seis miembros (6). Los anillos de cinco miembros también tienen menos libertad conformacional que los anillos de seis miembros.
Las sondas de espín pueden dirigirse a ciertos entornos en sistemas heterogéneos mediante la elección de los sustituyentes apropiados\(\mathrm{R}\) (Fig. 10.1). Las especies no sustituidas\((\mathrm{R}=\mathrm{H})\) son hidrofóbicas y se dividen preferiblemente en ambientes no polares. La preferencia por los aceptores de enlaces de hidrógeno se logra mediante derivados de hidroxilo\((\mathrm{R}=\mathrm{OH})\), mientras que los entornos iónicos pueden ser abordados por un grupo carboxilato a suficientemente alto\(\mathrm{pH}\left(\mathrm{R}=\mathrm{COO}^{-}\right)\) o por un grupo trimetilamonio\((\mathrm{R}=\)\(\left.\mathrm{N}\left(\mathrm{CH}_{3}\right)_{3}^{+}\right)\). Se\(\mathrm{R}\) utilizan grupos reactivos para SDSL, como el grupo metanotiosulfonato en el derivado dehidro-proxilo MTSL 5, el cual reacciona selectivamente con grupos tiol en condiciones suaves. Los grupos tiol se pueden introducir en las proteínas mediante mutación puntual dirigida al sitio de un aminoácido a cisteína y a ARN mediante el reemplazo de una nucleobase por tiouridina. En los derivados de DOXYL\(\mathbf{4}\), un anillo de seis miembros está espiro-unido a una cadena alquílica, que puede ser parte del ácido esteárico o de moléculas lipídicas. El\(\mathrm{N}^{\circ} \mathrm{O}^{\bullet}\) grupo en los derivados de DOXYL está unido rígidamente a la cadena alquílica y casi paralelo al eje de una hipotética cadena todo-trans.
El espectro de EPR de Nitróxido
A una buena aproximación, el sistema de espín de un radical nitróxido puede considerarse como un espín electrónico\(S=1 / 2\) acoplado al espín nuclear\(I=1\) del\({ }^{14} \mathrm{~N}\) átomo del\(\mathrm{N}-\mathrm{O}^{\bullet}\) grupo. Hiperfina
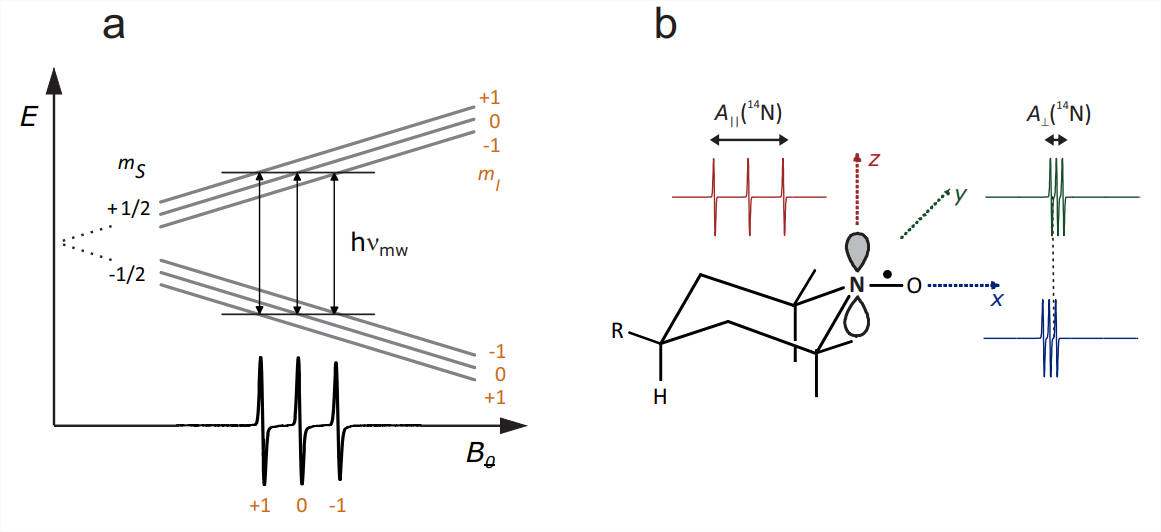
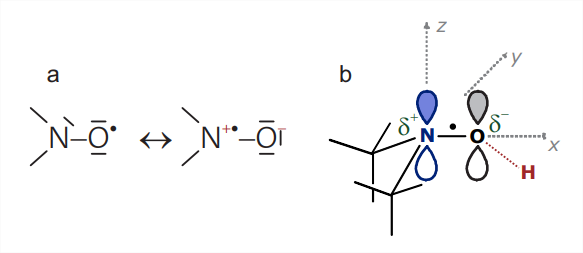
los acoplamientos a otros núcleos, como los protones de metilo, no suelen resolverse y contribuyen únicamente al ensanchamiento de la línea. El acoplamiento hiperfino al\({ }^{14} \mathrm{~N}\) átomo\(s p^{2}\) hibridado tiene una contribución significativa isotrópica de contacto de Fermi a partir de la densidad de espín en la\(2 s\) órbita y una contribución anisotrópica significativa de la densidad de espín en el\(p_{\pi}\) orbital que se combina con un\(p_{\pi}\) orbital en el átomo de oxígeno para dar al enlace N-O carácter de doble enlace parcial. La dirección de los lóbulos de la\(p_{\pi}\) órbita se elige como\(z\) eje molecular (Fig. 10.2 (b)). El tensor\({ }^{14} \mathrm{~N}\) hiperfino tiene simetría casi axial con\(z\) ser el eje único. El acoplamiento hiperfino es mucho más grande a lo largo\(z\) (del orden de\(90 \mathrm{MHz}\)) que en el\(x y\) plano (en el orden de\(15 \mathrm{MHz}\)).
El acoplamiento espín-órbita, que induce\(g\) anisotropía, surge principalmente en el\(\mathrm{O}\) átomo, donde un nivel de energía de par solitario está muy cerca del SOMO. El\(g\) tensor es ortorrómbico con asimetría casi máxima. El mayor\(g\) desplazamiento es positivo y se observa a lo largo del enlace N-O, que es el\(x\) eje del marco molecular\(\left(g_{x} \approx 2.009\right)\). Se observa un\(g\) desplazamiento intermedio a lo largo del\(y\) eje\(\left(g_{y} \approx 2.006\right)\), mientras que el\(g_{z}\) valor es muy cercano a\(g_{e}=2.0023\). A frecuencias de banda X, donde la\(\nu_{\mathrm{mw}} \approx 9.5 \mathrm{GHz}, g\) anisotropía corresponde solo a\(1.13 \mathrm{mT}\) dispersión en campos de resonancia, mientras que la anisotropía hiperfina corresponde a\(6.5 \mathrm{mT}\) dispersión. A frecuencias de banda W, donde\(\nu_{\mathrm{mw}} \approx 95 \mathrm{GHz}\), la anisotropía hiperfina sigue siendo la misma pero la\(g\) anisotropía aporta una dispersión diez veces mayor de\(11.3 \mathrm{mT}\), que ahora domina.
El espectro CW EPR barrido por campo para una sola orientación se puede entender considerando la regla de selección de que el número cuántico magnético\(m_{S}\) del espín electrónico debe cambiar en 1, mientras que el número cuántico magnético\(m_{I}\) del espín\({ }^{14} \mathrm{~N}\) nuclear debe no cambiar. Por lo tanto, cada transición puede asignarse a un valor de\(m_{I}\). Porque\(I=1\) hay tres valores de este tipo,\(m_{I}=-1,0\), y 1 (Fig. \(10.2(\mathrm{a})\)). La frecuencia de microondas\(\nu_{\mathrm{mw}}\) es fija y la resonancia se observa en campos donde la energía del cuántico de microondas\(h \nu_{\mathrm{mw}}\) coincide con la energía de una transición.
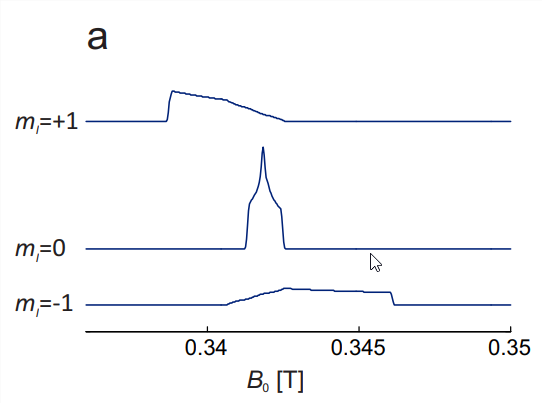
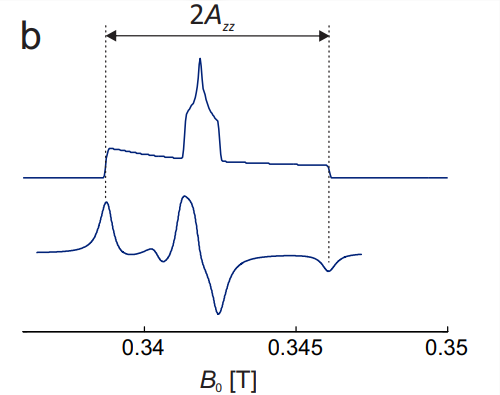
Para construir el espectro de estado sólido, se debe considerar la dependencia de orientación de las tres transiciones (Fig. 10.3 (a)). En cada orientación individual, la\(m_{I}=0\) línea es la línea central. Dado que las contribuciones hiperfinas escalan con\(m_{I}\), desaparece para esta línea y solo se observa\(g\) anisotropía. En la banda X, donde la anisotropía hiperfina domina con diferencia, esta línea es la más estrecha. El lineshape es el de\(g\) anisotropía pura (ver Fig. 3.4). Porque\(m_{I}=+1\), la orientación con el mayor\(g\) desplazamiento del campo de resonancia coincide con la del desplazamiento hiperfino más pequeño. De ahí que la dispersión de campo de resonancia menor por\(g\) anisotropía resta de la dispersión mayor por anisotropía hiperfina. Porque\(m_{I}=-1\), la situación es opuesta y las dos dispersiones se suman. De ahí que la\(m_{I}=-1\) transición, que en cualquier orientación dada es la línea de campo alto, tiene la mayor dispersión de resonancia, mientras que la\(m_{I}=+1\) transición de campo bajo tiene dispersión de campo de resonancia intermedia. La característica central en el espectro de absorción total (Fig. \(10.3(\mathrm{~b})\)) está fuertemente dominado por la\(m_{I}=0\) transición, mientras que los hombros externos corresponden a las transiciones\(m_{I}=+1\) (campo bajo) y\(m_{I}=-1\) (campo alto) en la\(z\) orientación. Por lo tanto, la división entre las extremidades externas en el espectro CW EPR, que corresponden a estos hombros en el espectro de absorción, es\(2 A_{z z}\).
Influencia de la dinámica en el espectro del nitróxido
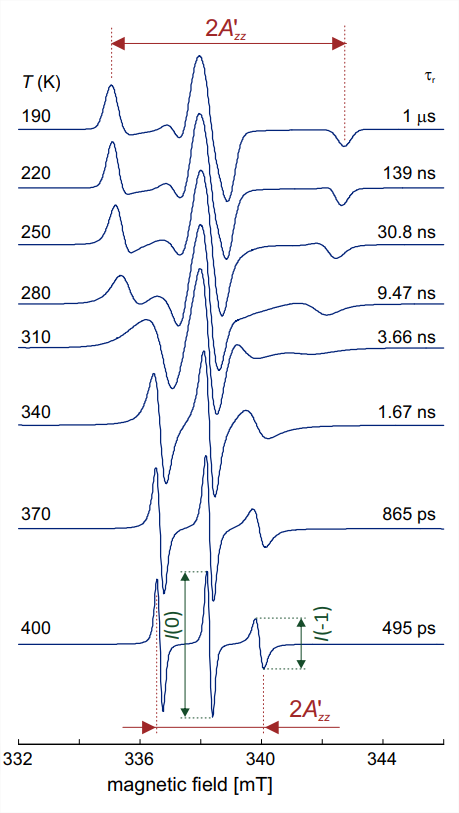
En solución líquida, las moléculas se tambalean estocásticamente debido a la difusión rotacional browniana. A continuación consideramos la difusión rotacional isotrópica, donde la molécula se tambalea con la misma velocidad promedio alrededor de cualquier eje en su marco molecular. Esta es una buena aproximación para sondas de espín de nitróxido con sustituyentes pequeños\(\mathrm{R}\). Por ejemplo, TEMPO\((\mathbf{1}\) con\(\mathrm{R}=\mathrm{H})\) es casi esférico con un radio de Van-der-Waals de\(3.43 \AA \AA\). En agua a temperatura ambiente, el tiempo de correlación\(\tau_{\mathrm{r}}\) rotacional para TEMPO es del orden de\(10 \mathrm{ps}\). El producto\(\tau_{\mathrm{r}} \Delta \omega\) con la anisotropía máxima\(\Delta \omega\) del espectro de nitróxido EPR en un eje de frecuencia angular es mucho menor que la unidad. En esta situación, se esperan promedios de anisotropía y tres líneas estrechas de igual anchura e intensidad. El espectro en la Fig. 10.2 (a) corresponde a esta situación y una mirada más cercana revela que la línea de campo alto tiene una amplitud algo menor. Esto se puede rastrear hasta un ancho de línea mayor que para las otras dos líneas, lo que indica una más corta\(T_{2}\) para la\(m_{I}=-1\) transición que para las otras transiciones. De hecho, la relajación transversal está dominada por el efecto de la combinación hiperfina y\(g\) anisotropía, que es mayor para la\(m_{I}=-1\) transición que tiene la mayor dispersión anisotrópica de frecuencias de resonancia. Con el aumento del tiempo de correlación rotacional\(\tau_{r}\), se espera que este proceso de relajación se vuelva más fuerte, lo que debería conducir a un mayor ensanchamiento de línea que sea más fuerte para la línea de campo alto y más débil para la línea central. Esto se observa de hecho en la simulación para\(\tau_{\mathrm{r}}=495\) ns mostrada en la traza inferior de la Fig. \(10.4\).
Según la teoría de relajación de Kivelson, la relación entre el ancho de línea de una de las líneas exteriores y el ancho de línea de la línea central viene dada por
\[\frac{T_{2}^{-1}\left(m_{I}\right)}{T_{2}^{-1}(0)}=1+B m_{I}+C m_{I}^{2}\]
donde
\[B=-\frac{4}{15} b \Delta \gamma B_{0} T_{2}(0) \tau_{\mathrm{r}}\]
y
\[C=\frac{1}{8} b^{2} T_{2}(0) \tau_{\mathrm{r}}\]
con el parámetro de anisotropía hiperfina
\[b=\frac{4 \pi}{3}\left[A_{z z}-\frac{A_{x x}+A_{y y}}{2}\right]\]
y el parámetro de anisotropía de Zeeman del electrón\(\Delta \gamma\)
\[\Delta \gamma=\frac{\mu_{\mathrm{B}}}{\hbar}\left[g_{z z}-\frac{g_{x x}+g_{y y}}{2}\right]\]
El tiempo de relajación\(T_{2}(0)\) para la línea central se puede calcular a partir del ancho de línea pico a pico correspondiente en el dominio de campo\(\Delta B_{p p}(0)\) como
\[T_{2}(0)=\frac{2}{\sqrt{3} g_{\text {iso }} \mu_{\mathrm{B}} \Delta B_{\mathrm{pp}}(0)}\]
Así, las ecuaciones (10.1-10.3) pueden resolverse para el único desconocido restante\(\tau_{\mathrm{r}}\). En la práctica, las relaciones de amplitudes de línea pico a pico\(I\left(m_{I}\right)\) se analizan en lugar de las relaciones de ancho de línea, ya que se pueden medir con mayor precisión. La relación de ancho de línea está relacionada con la relación de amplitud\(I(0) / I(-1)\) (ver traza inferior en la Fig. 10.4) en un espectro de primera derivada por
\[\frac{T_{2}^{-1}\left(m_{I}\right)}{T_{2}^{-1}(0)}=\sqrt{\frac{I(0)}{I\left(m_{I}\right)}}\]
ya que la intensidad integral de la línea de absorción (doble integral de la forma de línea derivada) es la misma para cada una de las tres transiciones. El tiempo de correlación rotacional se puede determinar por lo tanto, por ejemplo,
\[\tau_{\mathrm{r}}=\frac{\sqrt{3}}{2 b}\left[\frac{b}{8}-\frac{4 \Delta \gamma B_{0}}{15}\right]^{-1} \frac{g_{\mathrm{iso}} \mu_{\mathrm{B}}}{\hbar} \Delta B_{\mathrm{pp}}(0)\left[\sqrt{\frac{I(0)}{I(-1)}}-1\right]\]
donde\(\Delta B_{0}\) es el ancho de línea pico a pico de la línea central. Esta ecuación se puede aplicar en el régimen de volteo rápido, donde las tres líneas individuales para\(m_{I}=-1,0\), y aún\(+1\) pueden ser claramente reconocidas y tienen la forma de líneas de absorción derivadas simétricas.
Para un volteo más lento con\(\tau_{\mathrm{r}}>1.5 \mathrm{~ns}\), la forma de la línea se vuelve más compleja y se acerca al límite rígido (espectro de estado sólido) aproximadamente\(\tau_{\mathrm{r}}=1 \mu \mathrm{s}\) (Fig. 10.4). Estas formas lineales se pueden simular considerando el intercambio multisitio entre diferentes orientaciones de la molécula con respecto al campo magnético. A diferencia del intercambio de dos sitios, que se discute en la parte de RMN del curso de conferencias (ver Sección 3 de las notas de la conferencia de RMN), no se pueden obtener expresiones cerradas para el intercambio multisitio. Sin embargo, podemos estimar la escala de tiempo donde las características espectrales son más amplias y los tiempos de relajación transversal son los más cortos. La coalescencia para el intercambio de dos sitios se observa en\(\Delta \Omega / k=2 \sqrt{2}\). Al sustituir\(k\)\(1 / \tau_{\text {r and }} \Delta \Omega\) por la anisotropía máxima de\(7.6 \mathrm{mT}\), correspondiente a\(213 \mathrm{MHz}\), encontramos un “tiempo de coalescencia”\(2 \sqrt{2} / \Delta \Omega \approx 2.1\) ns. Las simulaciones en la Fig. \(10.4\)muestran de hecho que alrededor de este tiempo de correlación rotacional,
el carácter del espectro cambia de intercambio rápido de orientación (espectro de tipo líquido con tres picos distintos) a intercambio de orientación lento (espectro de tipo sólido).
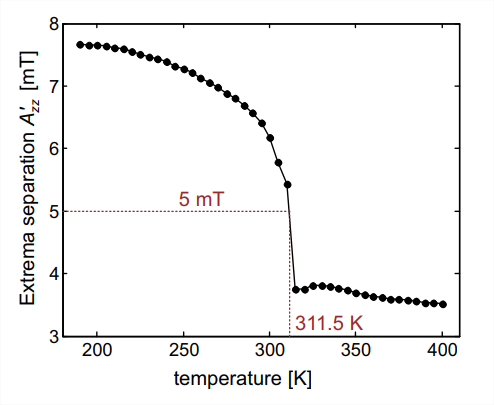
Una forma sencilla de analizar una dependencia de la temperatura, como la que se muestra en la Fig. 10.4, es graficar la separación\(2 A_{z z}^{\prime}\) extrema externa en función de la temperatura (Fig. 10.5). El “tiempo de coalescencia” en dicha parcela corresponde al gradiente más grande\(\mathrm{d} A_{z z}^{\prime} / \mathrm{d} T\), el cual coincide con la media entre\(2 A_{z z}^{\prime}\) los valores en el límite de volteo rápido y el límite rígido, que es\(5 \mathrm{mT}\). En el caso que nos ocupa, este tiempo de coalescencia es\(3.5 \mathrm{~ns}\) y se observa a una temperatura\(T_{5 \mathrm{mT}}=312\)\(\mathrm{K}\). La\(T_{5 \mathrm{mT}}\) temperatura es la temperatura donde el material se vuelve “blando” y las conformaciones moleculares pueden reorganizarse. Los espectros de nitróxido en el régimen de volteo lento pueden revelar más detalles sobre la dinámica, por ejemplo, si hay ejes de rotación preferidos, si el movimiento está restringido debido a la unión covalente del nitróxido a una molécula grande, o si hay orden local, como en una bicapa lipídica.
Polaridad y proticidad
La deslocalización del electrón desapareado en el\(\mathrm{N}-\mathrm{O}^{\bullet}\) grupo se puede entender considerando estructuras mesoméricas (Fig. 10.6). Si el electrón desapareado reside en el oxígeno, el número formal de electrones de valencia es cinco en nitrógeno y seis en oxígeno, lo que corresponde a la carga nuclear que no es compensada por electrones de capa interna. De ahí que ambos átomos sean formalmente neutros en esta estructura limitante. Si, por otro lado, el electrón desapareado reside en el átomo de nitrógeno, solo se asignan cuatro electrones de valencia a este átomo, mientras que siete electrones de valencia se asignan al átomo de oxígeno. Esto corresponde a la separación de carga con la carga positiva formal sobre nitrógeno y la carga negativa formal sobre oxígeno. La forma de carga separada se ve favorecida en los disolventes polares, los cuales filtran la atracción de Coulomb entre las dos cargas, mientras que la forma neutra se ve favorecida en los disolventes no polares. Por lo tanto, para un radical nitróxido dado en una serie de disolventes, se espera que el acoplamiento\({ }^{14} \mathrm{~N}\) hiperfino, que proviene de la densidad de espín en el átomo de nitrógeno, aumente con el aumento de la polaridad del disolvente. Este efecto efectivamente se ha encontrado. Se ve más fácilmente en el estado sólido para\(A_{z z}\) pero también se puede discernir en el estado líquido para\(A_{\text {iso }}\).
Se espera que el cambio en\(A_{z z}\) esté anti-correlacionado con el\(g_{x x}\) desplazamiento, debido a que este desplazamiento surge del SOC en el átomo de oxígeno y, cuanto mayor es la densidad de espín en el átomo de nitrógeno, menor es en el átomo de oxígeno. Este efecto también se ha encontrado y es más fácilmente detectado por EPR de campo alto/alta frecuencia a frecuencias de frecuencias de banda W de\(\approx 95 \mathrm{GHz}\) o incluso frecuencias más altas. \(A_{z z}\)La forma en que se correlaciona\(g_{x x}\) depende de la proticidad del solvente. Los solventes próticos forman enlaces de hidrógeno con los pares solitarios en el átomo de oxígeno del\({ }^{\bullet}\) grupo N-O. Esto disminuye la energía de los orbitales de pares solitarios, haciendo menos probable la excitación de un electrón de estos orbitales al SOMO. Dado que esta excitación proporciona la principal contribución al SOC y, por lo tanto, al\(g_{x x}\) desplazamiento, el enlace de hidrógeno al oxígeno reduce el\(g_{x x}\) desplazamiento. Si dos nitróxidos tienen el mismo acoplamiento hiperfino\(A_{z z}\) en un ambiente aprótico y prótico,\(g_{x x}\) será menor en el ambiente prótico. Este efecto también se ha encontrado. En algunos casos fue posible discernir etiquetas de nitróxido con cero, uno y dos enlaces de hidrógeno por resolución de sus\(g_{x x}\) características en espectros CW EPR de banda W. Se han encontrado pendientes de\(-1.35 \mathrm{~T}^{-1}\)\(-2 \mathrm{~T}^{-1}\) para aprótico en ambientes próticos para la correlación entre\(A_{z z}\) y\(g_{x x}\) para MTSL en bacteriorodopsina marcada con espín en bicapas lipídicas [Ste+00].
Accesibilidad al agua
La polaridad y la proticidad son parámetros proxy para la accesibilidad al agua de sitios marcados por espín en proteínas. Otras dos técnicas proporcionan información complementaria. Primero, el agua puede ser reemplazada por agua deuterada y se puede medir la profundidad de modulación del deuterio ESEEM. Debido a la\(r^{-6}\) dependencia de la profundidad de modulación (ver Ec. (8.7)) la técnica es más sensible a los núcleos de deuterio en las proximidades de la etiqueta de espín. Siempre\(k \ll 1\) y cuando se sumen las contribuciones de profundidad de modulación de núcleos individuales, de modo que la profundidad de modulación de deuterio total es una medida para la concentración de deuterio local cercana a la etiqueta. Los datos se pueden procesar de una manera que elimine la contribución de los núcleos directamente unidos a hidrógeno. Estrictamente hablando, esta técnica mide la concentración no solo de protones de agua sino también de la de cualquier protón intercambiable cerca de la etiqueta, pero sólo en la medida en que estos protones intercambiables son accesibles al agua durante la preparación o medición de la muestra.
Una segunda técnica más directa que es aplicable a temperatura ambiente mide la señal de RMN protónica en función de la potencia de microondas irradiada con la frecuencia de microondas encendida resonante con la transición central de una etiqueta de giro de nitróxido. Dicha irradiación transfiere la polarización de espín electrónico a protones de agua por el efecto Overhauser. Esta polarización nuclear dinámica (DNP) de Overhauser es altamente específica para el agua, ya que depende críticamente de que la señal de RMN de protón del agua sea estrecha y de la rápida difusión del agua. En biomoléculas, la accesibilidad al agua de las etiquetas de espín es alta en las superficies expuestas al agua de proteínas solubles y de membrana y baja dentro de las proteínas y en las superficies expuestas a lípidos. Para los transportistas, la accesibilidad al agua puede cambiar con el estado en el proceso de transporte.
Accesibilidad al oxígeno
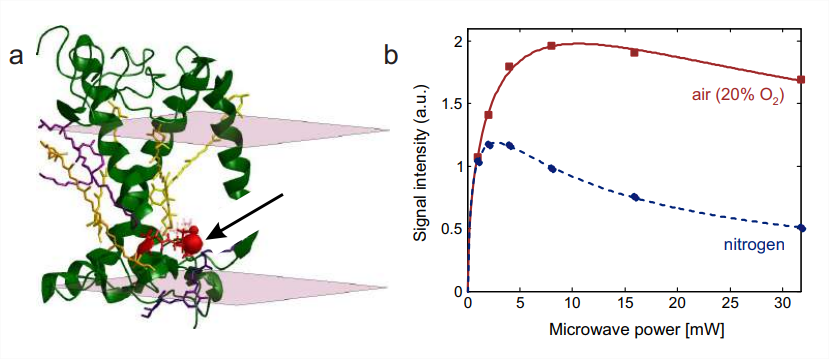
Dado que la colisión de oxígeno triplete paramagnético con sondas de espín mejora la relajación (Fig. \(7.4)\), el parámetro de saturación\(S=\omega_{1}^{2} T_{1} T 2\) es menor para las etiquetas giratorias accesibles al oxígeno que para las etiquetas giratorias no accesibles al oxígeno. Este cambio puede ser detectado por mediciones progresivas de saturación de potencia CW (Sección 7.2.2). El experimento se realiza de manera más conveniente con tubos capilares hechos del plástico permeable a los gases TPX. Se realiza una medición de referencia en atmósfera de nitrógeno, lo que provoca desoxigenación de la muestra en la escala de tiempo de 15 min. Luego se cambia la corriente de gas a aire (20% de oxígeno) u oxígeno puro y se repite la medición. Dichos datos se muestran en la Fig. \(10.7\)para el residuo 229 en el complejo de cosecha ligera de plantas principales LHCII. Este residuo está expuesto a lípidos. Como molécula no polar, el oxígeno se disuelve bien en la región de la cadena alquílica de una bicapa lipídica. En consecuencia, la señal se satura a mayor potencia en una atmósfera de aire que en una atmósfera de nitrógeno. La accesibilidad al oxígeno puede cuantificarse mediante un\(P_{1 / 2}\) parámetro normalizado (Sección 7.2.2).
Mediciones locales de pH
El acoplamiento\({ }^{14} \mathrm{~N}\) hiperfino de las sondas de espín de nitróxido se vuelve\(\mathrm{pH}\) sensible si el heterociclo que contiene el\(\mathrm{N}-\mathrm{O}^{\bullet}\) grupo también contiene un átomo de nitrógeno que puede protonarse en el\(\mathrm{pH}\) intervalo deseado. Esto se aplica, por ejemplo, al nitróxido de imidazolidina 3 de la Fig. \(10.1\), que tiene un valor de pK\(\approx 4.7\) y presenta un cambio en el acoplamiento isotrópico de\({ }^{14}\) N hiperfina\(0.13 \mathrm{mT}\) entre la\((1.56 \mathrm{mT})\) forma protonada (1.43 mT) y desprotonada, que puede resolverse fácilmente en líquido solución. Al modificar la sonda a un marcador, se\(\mathrm{pH}\) puede medir local cerca de un residuo de interés en una proteína.