2.1: Preludio a la Teoría Orbital Molecular
- Page ID
- 72031

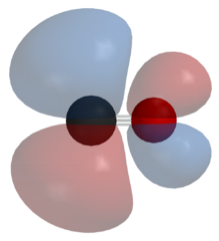
La teoría del enlace de valencia (VB) nos dio una imagen cualitativa de la unión química, la cual fue útil para predecir las formas de las moléculas, las fuerzas de enlace, etc., falla en describir algunas situaciones de unión con precisión porque ignora la naturaleza de onda de los electrones. La teoría orbital molecular (MO) tiene el potencial de ser más cuantitativa. Con él también podemos obtener una imagen de dónde están los electrones en la molécula, como se muestra en la imagen de la derecha. Esto puede ayudarnos a comprender patrones de unión y reactividad que de otra manera son difíciles de explicar.
Aunque la teoría MO en principio nos da una manera de calcular las energías y las funciones de onda de los electrones en las moléculas con mucha precisión, generalmente nos conformamos con modelos simplificados aquí también. Estos modelos simples no dan energías orbitales y de enlace muy precisas, pero sí explican conceptos como la resonancia (por ejemplo, en la molécula de ferroceno) que son difíciles de representar de otra manera. Podemos obtener energías más precisas de la teoría MO mediante el “cálculo numérico” computacional. Si bien la teoría MO es más correcta que la teoría VB y puede ser muy precisa en la predicción de las propiedades de las moléculas, también es bastante complicada incluso para moléculas bastante simples. Por ejemplo, no debería tener problemas para dibujar los cuadros VB para CO, NH 3 y benceno, pero encontraremos que estos son cada vez más desafiantes con la teoría de MO.