
5.12: Reacciones de sustitución de ligandos
- Page ID
- 72333

Los complejos de metales de transición pueden intercambiar un ligando por otro, y estas reacciones son importantes en su síntesis, estereoquímica y química catalítica. Los mecanismos de las reacciones químicas están íntimamente conectados con la cinética de reacción. Al igual que en la química orgánica, los mecanismos de las reacciones de metales de transición se deducen típicamente de experimentos que examinan la dependencia de la concentración de los ligandos entrantes y salientes en la velocidad de reacción, la detección de intermedios y la estereoquímica de los reactivos y productos.
Termodinámica vs. cinética. Cuando pensamos en las reacciones de los complejos de metales de transición, es importante recordar la distinción entre su termodinámica y cinética. Tomemos por ejemplo la formación del complejo de tetracyanonickelato plano cuadrado:
\[\ce{Ni^{2+}_{(aq)} + 4CN^{-}_{(aq)} = [Ni(CN)4]^{2-}_{(aq)}} \: \: \: K_{(eq)} \approx 10^{30} M^{-4}\]
Termodinámicamente, [Ni (CN) 4] 2- es muy estable, lo que significa que el equilibrio anterior se encuentra muy lejos a la derecha. Cinéticamente, sin embargo, el complejo es lábil, lo que significa que puede intercambiar sus ligandos rápidamente. Por ejemplo, el intercambio entre un ion CN marcado con 13 C y un ligando CN unido se produce en la escala de tiempo de decenas de milisegundos:
\[\ce{[Ni(CN)4]^{2-}_{(aq)} + *CN^{-}_{(aq)} -> [Ni(CN)3(*CN)]^{2-} + CN^{-}_{(aq)}} \: \: k_{exchange} \approx 10^{2}M^{-1}s^{-1}\]
Por el contrario, un compuesto puede ser termodinámicamente inestable pero cinéticamente inerte, lo que significa que tarda un tiempo relativamente largo en reaccionar. Por ejemplo, el ion [Co (NH 3) 6] 3+ es inestable en ácido, pero su reacción de hidrólisis con HCl concentrado tarda aproximadamente una semana en completarse a temperatura ambiente:
\[\ce{[Co(NH3)6]^{3+}_{(aq)} + 6H3O^{+}_{(aq)} -> [Co(H2O)6]^{3+}_{(aq)} + 6NH4^{+}_{(aq)}} \: \: K_{eq} \approx 10^{30}\]
Henry Taube, quien estudió los mecanismos de las reacciones de intercambio de ligandos en experimentos con tubos de ensayo simples, clasificó los complejos de metales de transición como lábiles si su semivida de reacción era de un minuto o menos, e inertes si tardaban más en reaccionar. El rango dinámico de tasas de sustitución de ligandos es enorme, abarcando al menos 15 órdenes de magnitud. En la escala temporal de la mayoría de los experimentos de laboratorio, la definición de labilidad Taube es útil para clasificar las reacciones en aquellas que tienen energías de activación bajas y altas. Como veremos, la energía de estabilización del campo cristalino (CFSE) juega un papel clave en la determinación de la energía de activación y por lo tanto la tasa de sustitución del ligando.
|
Henry Taube (Universidad de Stanford) recibió el Premio Nobel de 1983 por su trabajo sobre las reacciones de transferencia de electrones e intercambio de ligandos de complejos de metales de transición |

Energía de estabilización de campo cristalino y tasas de intercambio de ligandos. Consideremos una reacción de intercambio de ligandos muy común y simple, que es la sustitución de una molécula de agua por otra en un complejo octaédrico [M (H 2 O) 6] n+. Dado que los productos (excepto el marcador) son los mismos que los reactivos, sabemos que ΔG° = 0 y K eq = 1 para esta reacción. El progreso de la reacción puede controlarse por RMN usando agua marcada isotópicamente (que contiene típicamente 17 O o 18 O):
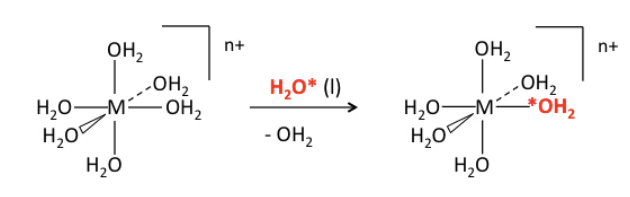
Lo más llamativo de esta reacción (de otra manera aburrida) es la gran diferencia en las constantes de velocidad -alrededor de 14 órdenes de magnitud- para diferentes iones metálicos y estados de oxidación:
| M n+ | log k (seg -1) |
|---|---|
| Cr 3+ |
|
| V 2+ |
|
| Cr 2+ |
|
| Cu 2+ |
|
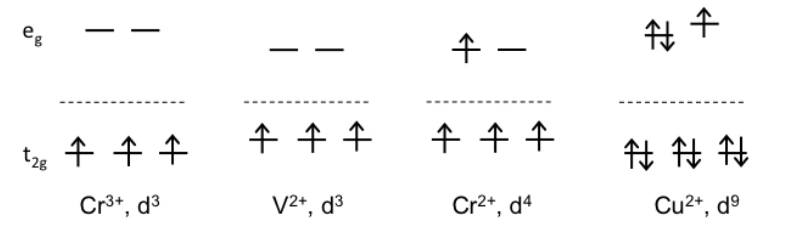
Si bien al principio puede parecer extraño que el mismo ion en dos estados de oxidación diferentes (Cr 3+ vs. Cr 2+) sea inerte o lábil, respectivamente, podemos comenzar a racionalizar la diferencia dibujando diagramas de división d-orbitales para los complejos. Lo que encontramos es que los complejos octaédricos que tienen un alto CFSE (Cr 3+, V 2+) tienden a ser inertes. Por el contrario, los iones con electrones en orbitales e g de alta energía (Cr 2+, Cu 2+) tienden a ser lábiles. En el caso de Cr 3+ y V 2+, la penalización de energía por distorsionar el complejo lejos de la simetría octaédrica -para hacer, por ejemplo, un intermedio de 5- o 7 coordenadas- es particularmente alta. Esta energía de activación para la sustitución de ligandos es menor para Cr 2+ y Cu 2+, que ya tienen electrones en orbitales antienlace e g.
Con base en las reglas que desarrollamos para calcular el CFSE de complejos de metales de transición, ahora podemos predecir las tendencias en las tasas de sustitución de ligandos:
- Complejos octaédricos con configuraciones d 3 y d 6 (espín bajo), como Cr 3+ (d 3), Co 3+ (d 6), Rh 3+ (d 6), Ru 2+ (d 6) y Os 2+ (d 6) tienden a ser inertes a la sustitución debido a su alto CFSE.
- Los complejos d 8 planos cuadrados, especialmente los de las series 4d y 5d, también son inertes a la ubstitución. Ejemplos son los complejos de Pd 2+, Pt 2+ y Au 3+.
- Los casos intermedios son complejos de Fe 3+, V 3+, V 2+, Ni 2+, y de iones del grupo principal (Be 2+, Al 3+) que son ácidos de Lewis duros. Estos complejos hacen fuertes enlaces metal-oxígeno y tienen tasas de cambio de agua en el rango de 10 1 -10 6 s -1.
- Los iones con cero CFSE intercambian moléculas de agua en una escala de tiempo de nanosegundos (k ≈ 10 8 -10 9 s -1). Estos incluyen iones con d 0, d 5 (alto espín) y d 10 recuentos de electrones, incluyendo metales alcalinos (Li +, Na +, K +, Rb +, Cs +) y alcalintérreos (Mg 2+, Ca 2+, Sr 2+, Sr 2+, Ba 2+), Zn 2+, Cd 2+, Hg 2+ y Mn 2+. En estos casos el CFSE es cero y el costo energético de romper la simetría octaédrica es relativamente bajo.
- Para los elementos del bloque p, se produce un intercambio más rápido con iones más grandes (por ejemplo, Ba 2+ > Ca 2+ y Ga 3+ > Al 3+), porque la fuerza ácida de Lewis disminuye al aumentar el tamaño de los iones.
- El ion Cu 2+ (d 9), como ion Jahn-Teller, ya está distorsionado lejos de la simetría octaédrica y por lo tanto es bastante lábil, intercambiando ligandos de agua a una velocidad de aproximadamente 10 8 s -1.
Mecanismos de sustitución de ligandos. Para un complejo ML n sometido a sustitución de ligando, existen esencialmente tres mecanismos de reacción diferentes:
- En el mecanismo disociativo, un complejo ML n primero pierde un ligando para formar un intermedio ML n-1, y el ligando entrante Y reacciona con el fragmento ML n-1:
\[\ce{L_{n-1}M-L <=>[-L, k_{1}][+L, k_{-1}] L_{n-1}M-\Box ->[+Y, k_{2}] L_{n-1}M-Y}\]
Este mecanismo se ilustra a continuación para la sustitución de ligando en un complejo octaédrico ML 6. El estado intermedio en este ejemplo involucra un fragmento trigonal bipiramidal ML 5 así como ligandos L e Y libres.
Si el paso determinante de la velocidad es la disociación de L del complejo, entonces la concentración de Y no afecta la velocidad de reacción, lo que lleva a la ley de velocidad de primer orden:
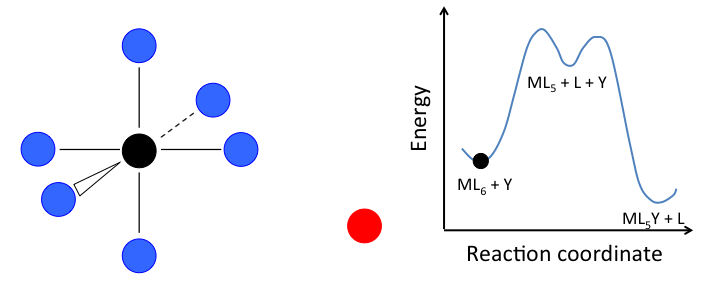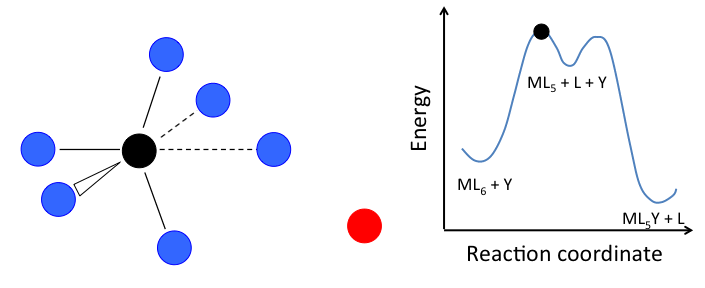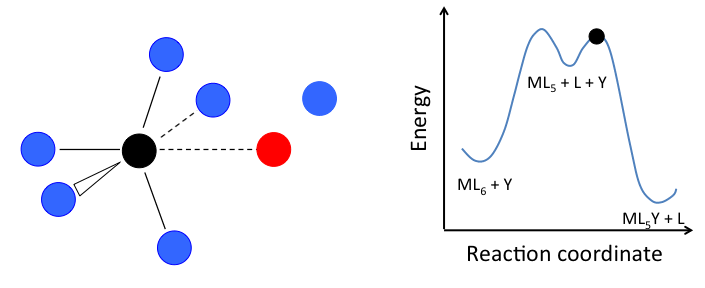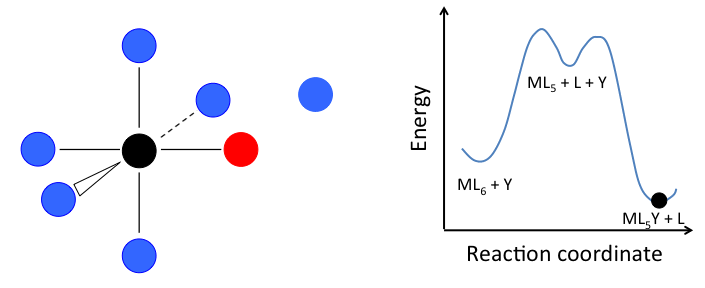
\[Rate=k_{1}[ML_{n}]\]
En el caso de un complejo octaédrico, esta reacción sería de primer orden en ML 6 y orden cero en Y, pero sólo si el estado de transición de energía más alto es el que precede a la formación del intermedio ML 5. Si los dos estados de transición están cerca en energía (como en el caso de la animación a la derecha), entonces la ley de tarifas se vuelve más complicada. En este caso, podemos simplificar el problema asumiendo una baja concentración en estado estacionario del intermedio ML n. La ley de tasas resultante es:
\[Rate= \frac{k_{1}k_{2}[Y][ML_{n}]}{k_{-1}[L] + k_{2}[Y]}\]
lo que reduce a la ley de tasa de primer orden más simple cuando k 2 [Y] >> k -1 [L]. Debido a que la formación del estado de transición implica la disociación de un ligando, la entropía de activación siempre es positiva en el mecanismo disociativo.
- En el mecanismo asociativo, el ligando entrante Y ataca el complejo ML n, formando transitoriamente un intermedio ML n Y, y el intermedio pierde entonces un ligando L formando el producto ML n-1 Y
Los complejos que experimentan sustitución asociativa son típicamente insaturados coordinadamente o contienen un ligando que puede cambiar su unión al metal, por ejemplo, un cambio en la aptitud o flexión de un ligando de óxido nítrico (NO). En catálisis homogénea, la vía asociativa es deseable porque el evento de unión, y por lo tanto la selectividad de la reacción, depende no solo de la naturaleza del catalizador metálico sino también de la molécula que está involucrada en el ciclo catalítico.
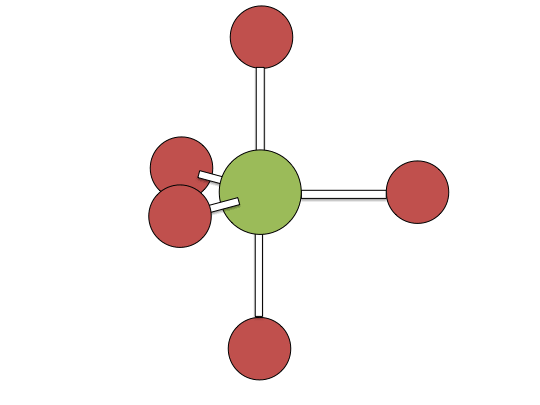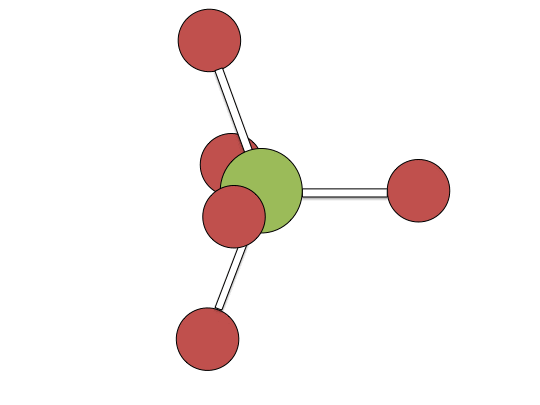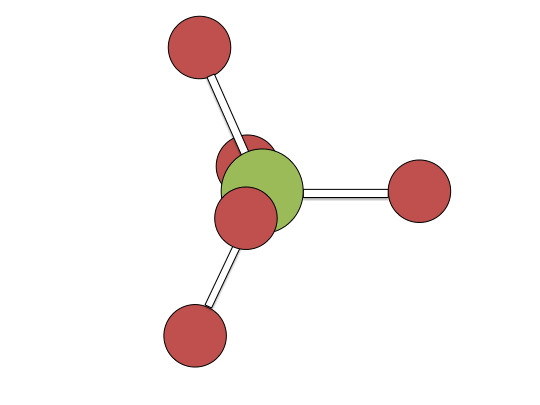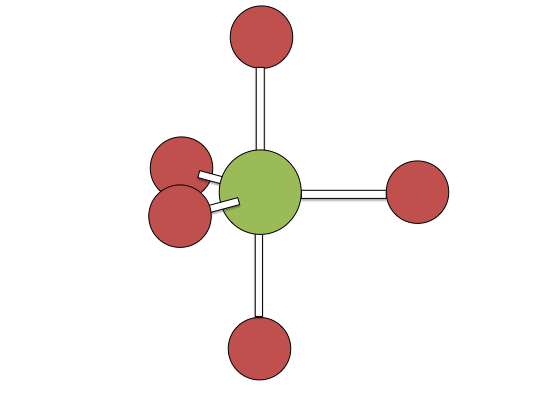
Mecanismo de pseudorotación de bayas
Ejemplos de mecanismos asociativos se encuentran comúnmente en la química de complejos metálicos planos cuadrados d 8, por ejemplo, el complejo de Vaska (IrCl (CO) [P (C 6 H 5) 3] 2) y tetracloroplatinato (II). Estos compuestos (ML 4) se unen al ligando entrante (sustitutivo) Y para formar intermedios pentacoordinados ML 4 Y, que en una etapa posterior disocian uno de sus ligandos. Aunque el ligando entrante se une inicialmente en un sitio ecuatorial, la pseudorrotación de Berry proporciona una vía de baja energía para que todos los ligandos muestreen tanto los sitios ecuatoriales como axiales. La disociación del ligando debe ocurrir desde un sitio ecuatorial de acuerdo con el principio de reversibilidad microscópica. La disociación de Y no da como resultado ninguna reacción, pero la disociación de L da como resultado una sustitución neta, produciendo el complejo d 8 ML 3 Y. El primer paso es típicamente determinante de la velocidad. Así, la entropía de activación es negativa, lo que indica un incremento en el orden en el estado de transición. Las reacciones asociativas siguen una cinética de segundo orden: la velocidad de aparición del producto depende de la concentración tanto de ML 4 como de Y.
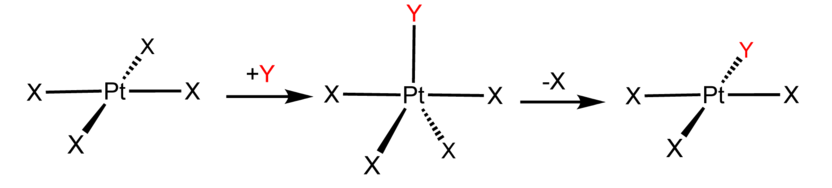
El Efecto Trans, que está conectado con el mecanismo asociativo, controla la estereoquímica de ciertas reacciones de sustitución de ligandos.
El efecto trans se refiere a la lestabilización (haciéndose más reactivos) de ligandos que son trans a ciertos otros ligandos, siendo estos últimos denominados ligandos trans-directores. La lestabilización de ligandos trans se atribuye a efectos electrónicos y es más notable en complejos planos cuadrados, pero también se puede observar con complejos octaédricos. [18] El efecto cis se observa con mayor frecuencia en los complejos octaédricos.
Además del efecto cinético trans, los ligandos trans también influyen en el estado fundamental de la molécula, siendo los más notables la longitud de los enlaces y la estabilidad. Algunos autores prefieren el término influencia trans para distinguirlo del efecto cinético, [19] mientras que otros utilizan términos más específicos como efecto trans estructural o efecto trans termodinámico. [18]
El descubrimiento del efecto trans se atribuye a Ilya Ilich Chernyaev, [20] quien lo reconoció y le dio nombre en 1926. [21]
La intensidad del efecto trans (medida por el incremento en la tasa de sustitución del ligando trans) sigue esta secuencia:
- F −, H 2 O, OH − < NH 3 < py < Cl − < Br − < I −, SCN −, NO 2 −, SC (NH 2) 2, Ph − < SO 3 2− < PR 3, ASr 3, SR 2, CH 3 − < H −, NO, CO, CN −, C 2 H 4
Tenga en cuenta que los ligandos de campo débil tienden a ser ligandos de dirección trans pobres, mientras que los ligandos de campo fuertes son fuertemente trans-dirigientes.
El ejemplo clásico del efecto trans es la síntesis del cisplatino y su isómero trans. [22] A partir de PtcL 4 2−, el primer ligando NH 3 se agrega al azar a cualquiera de las cuatro posiciones equivalentes. Sin embargo, dado que Cl − tiene un mayor efecto trans que NH 3, el segundo NH 3 se agrega trans a un Cl − y por lo tanto cis al primer NH 3.
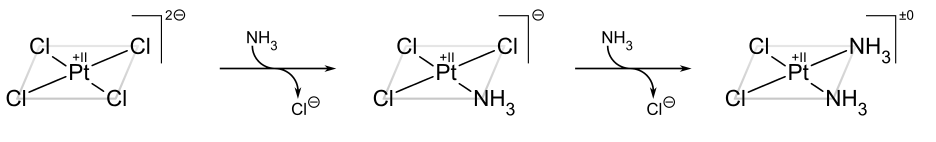
Si, por otro lado, uno parte de Pt (NH 3) 4 2+, se obtiene en su lugar el producto trans:
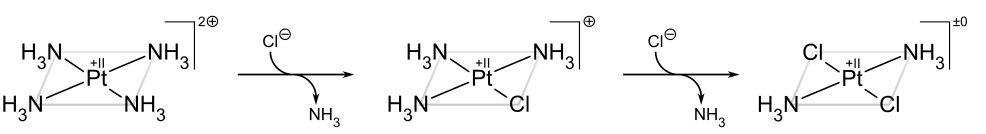
El efecto trans en complejos cuadrados puede explicarse en términos del mecanismo asociativo, descrito anteriormente, que pasa por un intermedio bipiramidal trigonal. Los ligandos con un alto efecto cinético trans son en general aquellos con alta acidez π (como en el caso de las fosfinas) o repulsiones de par solitario bajo ligando —d π (como en el caso del hidruro), que prefieren los sitios ecuatoriales más π-básicos en el intermedio. La segunda posición ecuatorial está ocupada por el ligando entrante. El tercer y último sitio ecuatorial está ocupado por el ligando trans saliente, por lo que el resultado neto es que el producto cinéticamente favorecido es aquel en el que se elimina el ligando trans al de mayor efecto trans. [19]
- El mecanismo de intercambio es similar a las vías asociativa y disociativa, excepto que no se forma ningún intermedio ML n Y o ML n-1 distinto. Este mecanismo concertado puede considerarse análogo a la sustitución nucleofílica a través de la vía S N 2 en un átomo de carbono tetraédrico en química orgánica. El mecanismo de intercambio se clasifica además como asociativo (I a) o disociativo (I d) dependiendo de la importancia relativa de la unión M-Y y M-L en el estado de transición. Si el estado de transición se caracteriza por la formación de un fuerte enlace M-Y, entonces el mecanismo es I a. Por el contrario, si el debilitamiento del enlace M-L es más importante para alcanzar el estado de transición, entonces el mecanismo es I d.
Un ejemplo del mecanismo I a es el intercambio de agua a granel y coordinada en [V (H 2 O) 6] 2+. En contraste, el ion ligeramente más compacto [Ni (H 2 O) 6] 2+ intercambia agua a través del mecanismo Id. [23]
Efectos del emparejamiento iónico. Los complejos catiónicos altamente cargados tienden a formar pares de iones con ligandos aniónicos, y estos pares de iones a menudo experimentan reacciones a través de la vía I a. El ligando entrante nucleofílico retenido electrostáticamente puede intercambiar posiciones con un ligando en la primera esfera de coordinación, dando como resultado una sustitución neta. Un proceso ilustrativo es la “anación” (reacción con un anión) del complejo hexaaquo de cromo (III):
\(\ce{[Cr(H2O)6]^{3+} + SCN^{-} <-> {[Cr(H2O)6], NCS}^{2+}}\)
\(\ce{{[Cr(H2O)6], NCS}^{2+} <-> [Cr(H2O)5NCS]^{2+} + H2O}\)