
14.1.1: Disociación y sustitución de ligandos
- Page ID
- 81628

Como se discute en el Capítulo 13, la sustitución de ligandos se caracteriza por un continuo de mecanismos unidos por extremos asociativo (A) y disociativo (D). En el extremo asociativo, el primer paso es una reacción asociativa; el ligando entrante forma un enlace con el metal y crea un intermedio de mayor número de coordinación. Entonces el ligando que sale toma su par solitario y se va. En el extremo disociativo, el primer paso es una reacción disociativa y el orden de los eventos es opuesto; se aparta un ligando y se forma un nuevo complejo con menor número de coordinación. Después, un nuevo ligando se asocia con el complejo en el segundo paso. A veces, en lugar de reaccionar, el “intermedio” es simplemente un complejo estable con un número de coordinación diferente al del reactivo.
Las reacciones más comunes que se utilizan para crear nuevos complejos son las disociaciones y sustituciones (que involucran tanto los pasos de disociación como de asociación). La sustitución asociativa es común para los complejos de 16 electrones (como\(d^8\) los complejos de Ni, Pd y Pt), mientras que tanto la disociación como la sustitución disociativa son la norma para los complejos de 18 electrones. Por otra parte, la realidad suele ser más complicada que estos extremos. En algunos casos, se dispone de evidencia para el intercambio (\(I_a\)o\(I_d\)).
En esta subsección, exploraremos las reacciones y mecanismos de sustitución de ligandos en el contexto de la química organometálica. Nos gustaría ser capaces de (a) predecir si un mecanismo es probable que sea asociativo o disociativo; (b) proponer un mecanismo razonable a partir de datos experimentales dados; y (c) describir los resultados que esperaríamos dado un mecanismo particular. Tenga en cuenta estos objetivos a medida que aprende las tuercas y tornillos teóricos y experimentales de las reacciones de sustitución.
Disociación
Se sabe que los compuestos de carbonilo experimentan reacciones de disociación al calentarse o irradiarse con luz. Por ejemplo, al calentarse, el dímero tricarbonilo de ciclopentadienilmolibdeno (\(\ce{Cp2Mo2(CO)6}\)), donde Cp es el ligando de ciclopentadienilo) pierde un ligando CO de cada ion Mo, y se forma un triple enlace entre los iones Mo (Top, Figura\(\PageIndex{1}\)). Un ejemplo de una disociación fotoactivada de CO es el caso del pentacarbonilo de hierro neutro,\(\ce{Fe(CO)5}\). La irradiación de\(\ce{Fe(CO)5}\) con UV produce\(\ce{Fe(CO)4}\) (Fondo, Figura\(\PageIndex{1}\)).
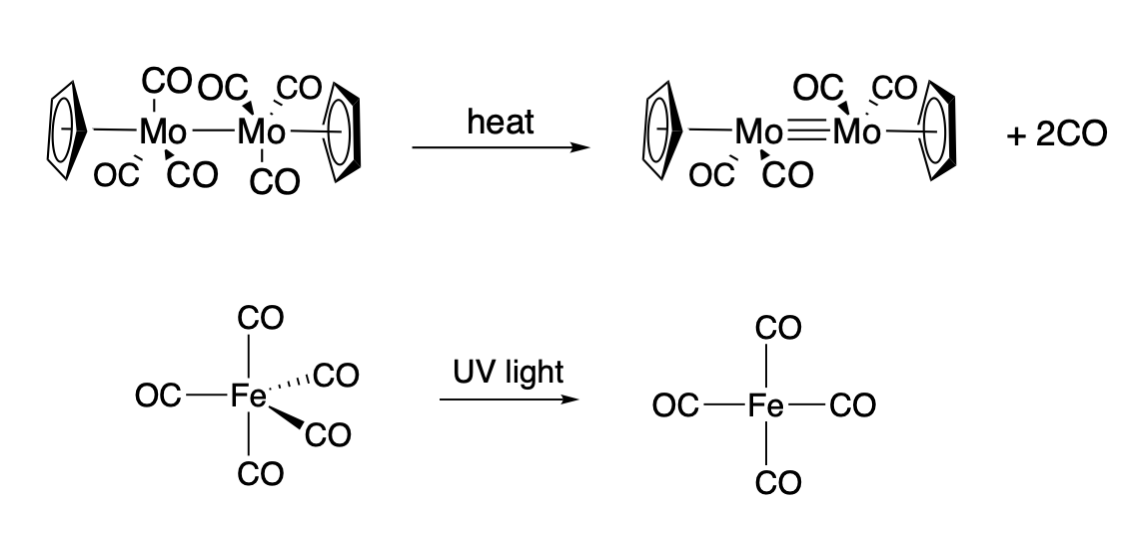
Sin embargo, en cada uno de los casos mostrados anteriormente, el complejo producto es reactivo hacia la asociación del ligando, generalmente dando como resultado una sustitución global. El dímero de ciclopentadienilmolibdeno dicarbonilo que se muestra en la parte superior de la Figura\(\PageIndex{1}\) se une a una variedad de sustratos a través del triple enlace metal-metal. Y el complejo de hierro tetracarbonilo que se muestra en la parte inferior en la Figura captura\(\PageIndex{1}\) fácilmente una variedad de ligandos para dar como resultado una sustitución global (ejemplo que se da a continuación); cuando\(\ce{Fe2(CO)9}\) se produce un ligando no asociado con él.
Fuentes: Artículos del dímero de tricarbonilo de pentacarbonilo de hierro y ciclopentadienilmolibdeno de Wikipedia.
Sustitución disociativa
La sustitución asociativa es poco probable para complejos saturados de 18 electrones; la coordinación de otro ligando produciría un intermedio de 20 electrones. Para los complejos de 18 electrones, los mecanismos de sustitución disociativa que involucran intermedios de 16 electrones son más probables. En un paso lento con entropía positiva de activación, el ligando de salida sale, generando un intermedio coordinativamente insaturado. El ligando entrante entra entonces en la esfera de coordinación del metal para generar el producto. Para el resto de este post, nos centraremos en la cinética de la reacción y la naturaleza del intermedio insaturado (que influye en la estereoquímica de la reacción). El reverso del primer paso, la re-coordinación del ligando saliente (constante de velocidad k—1), suele ser competitivo con la disociación.

Cinética de reacción
Empecemos por la situación general en la que\(k_1\) y\(k_{–1}\) son similares en magnitud. Dado que\(k_1\) es limitante de velocidad,\(k_2\) se supone que es mucho mayor que\(k_1\) y\(k_{–1}\). Lo más importante es que debemos asumir que la variación en la concentración del intermedio insaturado es esencialmente cero. Esto se llama aproximación de estado estacionario, y nos permite establecer una ecuación que relaciona la velocidad de reacción con concentraciones observables. Aferrarse a eso por un segundo; primero, podemos usar el paso 2 para establecer una expresión de velocidad preliminar.
\[\text{rate} = k_2[L_nM–◊][Li] \tag{1}\]
Por supuesto, el complejo insaturado está presente en concentraciones muy pequeñas y es inconmensurable, por lo que esta ecuación no nos ayuda mucho. Necesitamos eliminar la concentración del intermedio inconmensurable de (1), y la aproximación del estado estacionario nos ayuda a hacer esto. Podemos expresar variación en la concentración del intermedio insaturado como (procesos que lo hacen) menos (procesos que lo destruyen), multiplicándose por una duración de tiempo arbitraria para hacer que las unidades funcionen. Todo eso equivale a cero, según la aproximación SS. ¡La matemática dolorosa casi ha terminado! Dado que Δt no debe ser cero, el otro factor, la colección de términos, debe ser igual a cero.
\[ Δ[L_nM–◊] = 0 = (k1[L_nM–L^d] – k–1[L_nM–◊][L^d] – k_2[L_nM–◊][Li])Δt \tag{2}\]
\[0 = k_1[L_nM–Ld] – k_{-1}[L_nM–◊][Ld] – k_2[L_nM–◊][Li] \tag{3}\]
Reordenando para resolver\([L_nM–◊]\), llegamos a lo siguiente.
\[ [L_nM–◊] = k_1 \dfrac{[L_nM–L_d]}{(k_{-1}[L_d] + k_2[Li]} \tag{4}\]
Finalmente, sustituyendo en la ecuación (1) alcanzamos una ecuación de tasa verificable.
\[ \text{rate} = k_2k_1 \dfrac{[L_nM–Ld][Li]}{(k_{-1}[L_d] + k_2[Li]} \tag{5}\]
Cuando\(k_{–1}\) es insignificante, (5) se reduce a la ecuación familiar (6), típica de reacciones disociativas como S N 1.
\[\text{rate} = k_1[L_nM–L_d] \tag{6}\]
A diferencia de la ley de tasa asociativa, esta tasa no depende de la concentración de ligando entrante. Para reacciones que se describen mejor por (5), podemos ahogar la reacción en el ligando entrante para hacer\(k_2[Li]\) mucho mayor que\(k_{-1}[Ld]\), esencialmente forzando la reacción a ajustarse a la ecuación (6).
El intermedio insaturado y la estereoquímica
La disociación de un ligando de un complejo octaédrico genera un intermedio ML5 usaturado. Cuando los cinco ligandos restantes son de tipo L, como en Cr (CO) 5, el metal tiene 6 d electrones para un recuento total de electrones de 16. La geometría bipiramidal trigonal presenta problemas electrónicos (electrones desapareados) para electrones de 6 d, como muestra la siguiente figura. Los niveles de energía orbitales provienen de la teoría del campo cristalino. La distorsión a una pirámide cuadrada o una geometría de TBP distorsionada elimina el problema electrónico, por lo que los complejos d6 de cinco coordenadas suelen tener geometrías de TBP piramidales cuadradas o distorsionadas. ¡Esto es solo el proceso de predicción de geometría en acción!

Cuando el intermedio adopta geometría piramidal cuadrada (favorecida por buenos aceptores π-y σ-donadores... ¿por qué?) , el ligando entrante puede simplemente acercarse a donde se fue el ligando de salida, dando como resultado la retención de la estereoquímica. La inversión es más probable cuando el intermedio es una bipirámide trigonal distorsionada (favorecida por buenos donadores π). Como ya hemos visto para la sustitución asociativa, la fluxionalidad en el intermedio de cinco coordenadas puede complicar la estereoquímica de la reacción.
Fomentar la Sustitución Dissocativa
En general, la introducción de características estructurales que estabilizan el intermedio insaturado o desestabilizan el complejo de partida puede fomentar la sustitución disociativa. Ambas estrategias reducen la barrera de activación para la reacción. Otras formas extravagantes de fomentar la disociación incluyen métodos fotoquímicos, oxidación/reducción y abstracción de ligandos.
Comencemos con características que estabilizan el intermedio insaturado. Electrónicamente, al intermedio le encanta cuando su conteo de electrones d se adapta muy bien a sus orbitales de campo de cristal. A medida que estudie química organometálica, aprenderá que hay ciertos recuentos de electrones d “naturales” para geometrías particulares que encajan bien con los orbitales centrados en metales predichos por la teoría del campo de cristal. La geometría octaédrica es ideal para seis electrones d, por ejemplo, y la geometría plana cuadrada ama ocho electrones d. Los complejos con recuentos de electrones d “naturales”, pero con un ligando extra, están maduros para la sustitución disociativa. Los ejemplos clásicos son los complejos d8 TBP, que se convierten en complejos planos cuadrados d8 (piense en Pt (II) y Pd (II)) tras la disociación. Factores similares en realidad estabilizan los complejos iniciales de 18 electrones, haciéndolos menos reactivos en las reacciones de sustitución disociativa. Los complejos octaédricos d6 son particularmente felices y reaccionan más lentamente en sustituciones disociativas. Los tres tipos más comunes de complejos de 18 electrones, desde el más rápido hasta el más lento en la sustitución disociativa, son:
\(d^8\)TBP >\(d^10\) tetraédrico >\(d^6\) octaédrico
La desestabilización del complejo de partida se logra comúnmente añadiendo volumen estérico a sus ligandos. Naturalmente, la disociación alivia la congestión estérica en el complejo inicial. La quelación tiene el efecto contrario, y tiende a acerear el complejo de partida contra la disociación.

Planeo cubrir los métodos “extravagantes” en una publicación propia, pero estos incluyen estrategias como N-óxidos para la eliminación de CO, escisión fotoquímica del enlace ligando que sale del metal y el uso de cationes de plata para abstraer ligandos de haluro. La oxidación y reducción también pueden usarse para fomentar la sustitución: los complejos de 17 y 19 electrones son mucho más reactivos hacia la sustitución que sus análogos de 18 electrones.
Sustituciones asociativas
A pesar de la santidad de la regla de 18 electrones en la química organometálica, una amplia variedad de complejos estables poseen menos de 18 electrones totales en el centro del metal. Quizás los ejemplos más famosos de estos complejos son los complejos de 14 y 16 electrones de metales del grupo 10 (Ni, Pt, Pd). La sustitución de ligando en complejos de esta clase generalmente ocurre a través de un mecanismo asociativo, que implica el acercamiento del ligando entrante al complejo antes de la salida del grupo saliente. Si tenemos en mente este principio, parece bastante fácil predecir cuándo es probable que la sustitución del ligando sea asociativa. Pero, ¿cómo podemos detectar un mecanismo asociativo en los datos experimentales y cuáles son algunas de las consecuencias de este mecanismo?
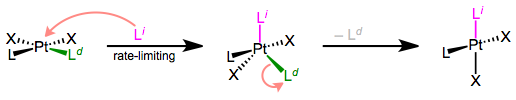
The prototypical mechanism of associative ligand substitution. The first step is rate-determining. A typical mechanism for associative ligand substitution is shown above. It should be noted that square pyramidal geometry is also possible for the intermediate, but is less common. Let’s begin with the kinetics of the reaction.
Reaction Kinetics for Associative Reactions
Reaction kinetics are commonly used to elucidate organometallic reaction mechanisms, and ligand substitution is no exception. Different mechanisms of substitution may follow different rate laws, so plotting the dependence of reaction rate on concentration often allows us to distinguish mechanisms. Associative substitution’s rate law is analogous to that of the SN2 reaction—rate depends on the concentrations of both starting materials.
\[ L_nM–L^d + L_i → L_nM–L_i + L^d \]
\[ \dfrac{d[L_nM–L^i]}{dt} = rate = k_1[L_nM–L^d][L^i] \]
The easiest way to determine this rate law is to use pseudo-first-order conditions. Although the rate law is second order overall, if we could somehow render the concentration of the incoming ligand unchanging, the reaction would appear first order. The observed rate constant under these conditions reflects the constancy of the incoming ligand’s concentration (\(k_{obs} = k_1[L^i]\), where both \(k_1\) and \([Li]\) are constants). How can we make the concentration of the incoming ligand invariant, you ask? We can drown the reaction in ligand to achieve this. The teensy weensy bit actually used up in the reaction has a negligible effect on the concentration of the “sea” of starting ligand we began with. The observed rate is equal to \(k_{obs}[L_nM–L^d]\), as shown by the purple trace below. By determining \(k_{obs}\) at a variety of \([L^i]\) values, we can finally isolate \(k_1\), the rate constant for the slow step. The red trace below at right shows the idea.
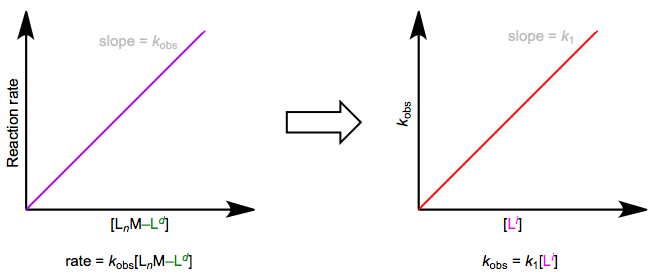
En muchos casos, el rastro rojo termina con una intercepción y distinta de cero... curioso, si nos limitamos al mecanismo simple que se muestra en la primera figura de este post. Una intercepción distinta de cero sugiere un mecanismo más complejo. Necesitamos agregar un nuevo término (llamado por razones\(k_s\) para aclararse en breve) a nuestro primer conjunto de ecuaciones:
\[rate = (k_1[L_i] + k_s)[L_nM–L^d]\]
\[k_{obs} = k_1[L_i] + k_s\]
La ley de tasa completa sugiere que algún otro paso (con tasa ks [LNM—ld]) independiente del ligando entrante está involucrado en el mecanismo. Para explicar esta observación, podemos invocar al disolvente como reactivo. El solvente puede asociarse con el complejo primero en una etapa lenta, luego el ligando entrante puede desplazar el solvente en una etapa rápida. La concentración de solvente no entra en la ley de tarifas porque, bueno, ¡está ahogando a los reactivos y su concentración sufre un cambio insignificante! A continuación se muestra un ejemplo de este mecanismo en el contexto de la química de Pt (II).
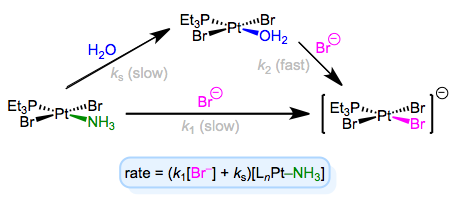
As an aside, it’s worth mentioning that the entropy of activation of associative substitution is typically negative. Entropy decreases as the incoming ligand and complex come together in the rate-determining step. Dissociative substitution shows the opposite behavior: loss of the departing ligand in the RDS increases entropy, resulting in positive entropy of activation.
Stereochemistry of Associative Substitution
As we saw in discussions of the trans effect, the entering and departing ligands both occupy equatorial positions in the trigonal bipyramidal intermediate. Microscopic reversibility is to blame: the mechanism of the forward substitution (displacement of the leaving by the incoming ligand) must be the same as the mechanism of the reverse reaction (displacement of the incoming by the leaving ligand). This can be a confusing point, so let’s examine an alternative mechanism that violates microscopic reversibility.
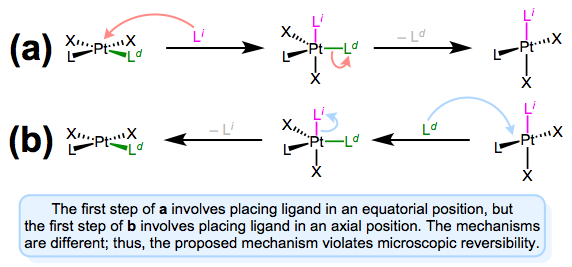
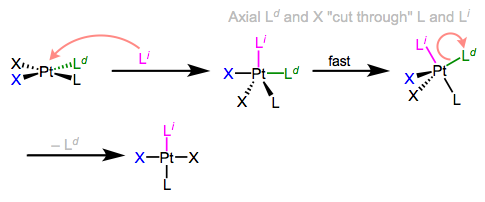
La figura anterior muestra por qué no es posible un mecanismo que implique aproximación axial y salida ecuatorial (o viceversa). Las reacciones hacia adelante y hacia atrás difieren, de hecho, en ambos pasos. En el mecanismo directo a, el ligando entrante entra en un sitio axial. Pero en la reacción inversa, el ligando entrante (es decir, el ligando de salida en el mecanismo a) se asienta en un sitio ecuatorial. Los segundos pasos de cada mecanismo también difieren: implica la pérdida de un ligando ecuatorial, mientras que b implica la pérdida de un ligando axial. En pocas palabras, este mecanismo viola la reversibilidad microscópica. ¿Y qué pasa con un mecanismo que implica aproximación axial y salida axial? Tal mecanismo es poco probable por motivos electrónicos. Los sitios ecuatoriales son más ricos en electrones que los sitios axiales, y se espera que la unión σ al\(d_{z^2}\) orbital axial sea fuerte. Intuitivamente, entonces, la pérdida de ligando de un sitio axial es menos favorable que la pérdida de un sitio ecuatorial.
Sé lo que estás pensando: ¿qué diablos tiene que ver todo esto con la estereoquímica? Observe que, en el mecanismo ecuatorial-ecuatorial (primera figura de este post), los ligandos axiales no se mueven en absoluto. La configuración del complejo de partida queda así retenida en el producto. Aunque la retención es “normal”, a menudo surgen complicaciones porque los complejos de TBP de cinco coordenadas, como otros complejos organometálicos de coordenadas impares, suelen ser fluxionales. Los ligandos axiales y ecuatoriales pueden intercambiar rápidamente a través de un proceso llamado pseudorrotación Berry, que se asemeja a los ligandos axiales “cortando” un par de ligandos ecuatoriales como tijeras (¡animación!). Fluxionalidad significa que todas las apuestas estereoquímicas están desactivadas, ya que cualquier ligando puede ocupar factiblemente un sitio ecuatorial. En el siguiente ejemplo, el ligando de partida comienza en cis a L, pero el ligando entrante termina en trans a L.
Dr. Michael Evans (Georgia Tech)
\(\PageIndex{6}\): Berry pseudorotation in the midst of associative ligand substitution. (Michael Evans)Associative Substitution in 18-electron Complexes?
Associative substitution can occur in 18-electron complexes if it’s preceded by the dissociation of a ligand. For example, changes in the hapticity of cyclopentadienyl or indenyl ligands may open up a coordination site, which can be occupied by a new ligand to kick off associative substitution. An allyl ligand may convert from its π to σ form, leaving an open coordination site where the π bond left. A particularly interesting case is the nitrosyl ligand—conversion from its linear to bent form opens up a site for coordination of an external ligand.
Summary of Associative Substitution
Associative ligand substitution is common for complexes with 16 total electrons or fewer. The reaction is characterized by a second-order rate law, the possibility of solvent participation, and a trigonal bipyramidal intermediate that is often fluxional. An open coordination site is essential for associative substitution, but such sites are often hidden in the dynamism of 18-electron complexes with labile ligands.
Quirky Substitutions
Over the years, a variety of “quirky” substitution methods have been developed. All of these have the common goal of facilitating substitution in complexes that would otherwise be inert. It’s an age-old challenge: how can we turn a stable complex into something unstable enough to react? Photochemical excitation, oxidation/reduction, and radical chains all do the job, and have all been well studied. We’ll look at a few examples in this post—remember these methods when simple associative or dissociative substitution won’t get the job done.
Photochemical Substitution
Substitution reactions of dative ligands—most famously, CO—may be facilitated by photochemical excitation. We just discussed the photochemically-activated dissociation of CO from ironpentacarbonyl above; that reaction is often used to accomplish substitution of a CO ligand. Other CO complexes also undergo photochemically-activated CO substitution reactions. Two examples are shown below. The first reaction yields only monosubstituted product without ultraviolet light, even in the presence of a strongly donating phosphine.

Todos los signos apuntan a mecanismos disociativos para estas reacciones (los complejos de partida tienen 18 electrones totales cada uno). La excitación, entonces, debe aumentar el carácter antiligante M-L de los electrones del complejo; exactamente cómo ocurre este aumento en el carácter antiligante ha sido un tema de cierto debate. Originalmente, la explicación predominante fue que el LUMO tiene carácter antienlace M-L, y la excitación patea un electrón desde el HOMO al LUMO, fomentando la escisión del enlace M—L. Una explicación más reciente y más sutil respaldada por cálculos respalda la participación de un estado de transferencia de carga de metal a ligando junto con el estado excitado de campo de ligando “clásico”.
Oxidación/Reducción
Imagínese a un bebé gritando sin su chupete, esa es una bonita analogía para un complejo organometálico de electrones impares. Los complejos que llevan 17 y 19 electrones totales son mucho más reactivos hacia la sustitución que sus homólogos de electrones pares. Por lo tanto, la oxidación y reducción de electrones simples (“sacar el chupete”, si se quiere) se puede utilizar para activar de manera eficiente la sustitución. Como cabría esperar, la oxidación y la reducción funcionan mejor en complejos ricos en electrones y pobres en electrones, respectivamente. El complejo de Mn en el ejemplo oxidativo a continuación, por ejemplo, incluye un grupo MeCP fuertemente donador (no mostrado).

La reducción funciona bien para los complejos carbonílicos metálicos pobres en electrones, que están felices de aceptar un electrón adicional.
Existe un método de oxidación de dos electrones que también vale la pena conocer: la oxidación del CO con óxidos de amina. Este pequeño método ingenioso libera dióxido de carbono, amina y un complejo insaturado que puede ser apagado por un ligando que cuelga alrededor. El truco es la adición al ligando de CO seguido de la eliminación del complejo insaturado. A medida que el CO2 oxidado y la amina reducida flotan, el complejo metálico encuentra otro ligando.

Procesos de Cadena Radical
La abstracción de átomos de complejos de 18 electrones produce intermedios neutros de 17 electrones, que son susceptibles a la sustitución de ligandos a través de mecanismos de cadena radical El hecho de que los intermedios sean neutros distingue estos métodos de los métodos basados en la oxidación. Los hidruros metálicos de primera fila son excelentes para estas reacciones, debido a sus enlaces M—H relativamente débiles. A continuación se muestra un ejemplo.

Después de la abstracción del átomo de hidrógeno por el iniciador, la sustitución es rápida y puede ocurrir varias veces. La propagación comienza de nuevo cuando el radical sustituido extrae hidrógeno del material de partida para regenerar el radical propagador y formar el producto. Estos métodos extravagantes son agradables de tener en tu bolsillo trasero cuando estás retrocedido en una esquina sintética; a veces, la sustitución asociativa y disociativa convencional simplemente no hará el trabajo. En el siguiente post, continuaremos con la adición oxidativa.