
8.8: Efectos Estructurales y Solventes en\(S_N\) Reactions
- Page ID
- 73191

Primero consideraremos la relación entre las estructuras de los derivados alquílicos y sus velocidades de reacción hacia un nucleófilo dado. Esto será seguido por una discusión de las reactividades relativas de varios nucleófilos hacia un derivado de alquilo dado. Por último, comentaremos con más detalle el papel del solvente en\(\text{S}_\text{N}\) las reacciones.
Estructura del Grupo Alquílico,\(R\), en\(\text{S}_\text{N}2\) Reacciones
Las tasas de reacciones\(\text{S}_\text{N}2\) de desplazamiento de los derivados alquílicos simples,\(RX\), siguen el orden primario\(R\)\(>\) secundario\(R\)\(\gg\) terciario\(R\). En síntesis prácticas que implican\(\text{S}_\text{N}2\) reacciones, los compuestos primarios generalmente funcionan muy bien, los isómeros secundarios son justos y los isómeros terciarios son casi completamente imprácticos. El impedimento estérico parece ser particularmente importante en la determinación de las velocidades de\(\text{S}_\text{N}2\) reacción, y la lentitud de los haluros terciarios parece estar mejor explicada por el obstáculo estérico al enfoque posterior de un nucleófilo atacante por los grupos alquilo en el carbono de reacción. Los datos pertinentes, que muestran cómo los grupos alquilo afectan la\(\text{S}_\text{N}2\) reactividad hacia el ión yoduro, se dan en la Tabla 8-1. Los grupos alquilo no solo suprimen la reactividad cuando están en el mismo carbono que el grupo lábil\(X\), como en el bromuro de terc- butilo, sino que también tienen efectos retardantes cuando se encuentran a un carbono lejos del grupo lábil. Esto es evidente en los datos del Cuadro 8-1 para 1-bromo-2,2-dimetilpropano (bromuro de neopentilo), el cual es muy poco reactivo en\(\text{S}_\text{N}2\) las reacciones. Los modelos a escala indican que el retraso es el resultado del impedimento estérico por los grupos metilo ubicados en el\(\beta\) carbono adyacente al nucleófilo que se aproxima:
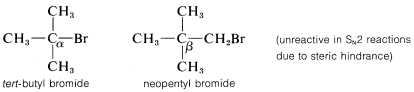
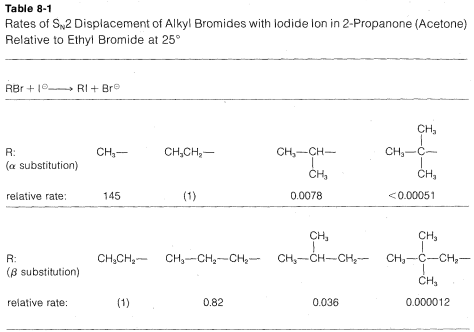
Además de los efectos estéricos, otros efectos estructurales de\(R\) influencia la\(\text{S}_\text{N}2\) reactividad de\(RX\). Un doble enlace\(\beta\) al halógeno,\(^6\) como en los cloruros de 2-propenilo, fenilmetilo (bencilo) y 2-oxopropilo mejora la reactividad de los compuestos hacia los nucleófilos. Así, las reactividades relativas hacia\(I^\ominus\) la 2-propanona son
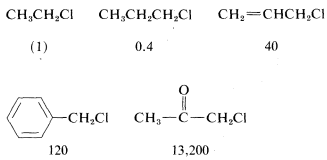
Las posibles razones de estas altas reactividades se discutirán más adelante (Sección 14-3B).
Estructura del Grupo Alquílico,\(R\), en\(\text{S}_\text{N}1\) Reacciones
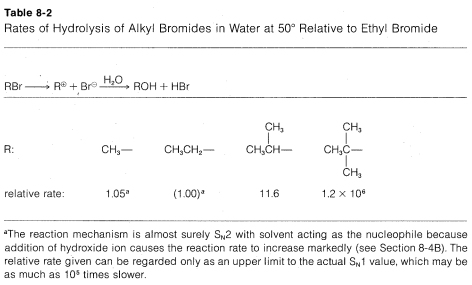
Las tasas de\(\text{S}_\text{N}1\) reacciones de los derivados alquílicos simples siguen el orden terciario\(R\)\(\gg\) secundario\(R\)\(>\) primario\(R\), que es exactamente opuesto al de\(\text{S}_\text{N}2\) las reacciones. Esto es evidente a partir de los datos del Cuadro 8-2, que enumera las velocidades relativas de hidrólisis de algunos bromidos de alquilo; solo los bromidos secundarios y terciarios reaccionan a velocidades medibles, y el bromuro terciario reacciona algunas\(10^5\) veces más rápido que el bromuro secundario.
¿Por qué los compuestos de alquilo terciario se ionizan mucho más rápidamente que los compuestos secundarios o primarios? La razón es que los cationes alquilo terciarios son más estables que los cationes secundarios o primarios y por lo tanto se forman más fácilmente. Esto lo apreciará mejor al observar el diagrama de energía de la Figura 8-4, que muestra el perfil de cambios energéticos para la hidrólisis de un compuesto alquílico,\(RX\), por el\(\text{S}_\text{N}1\) mecanismo. La tasa de
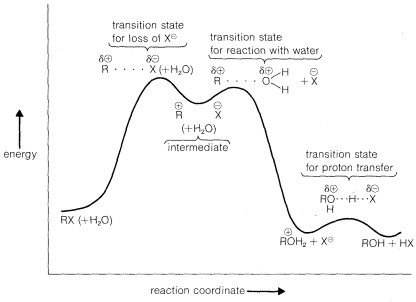
la reacción se determina por la etapa de ionización, o por la energía del estado de transición con respecto a la de los reactivos. En realidad, la energía del estado de transición es solo ligeramente superior a la energía de los intermedios iónicos\(R^\oplus X^\ominus\). Así, a una primera aproximación, podemos decir que la velocidad de ionización de\(RX\) dependerá de las energías de los iones formados. Ahora bien, si comparamos las tasas para una serie de compuestos\(RX\),, todos teniendo el mismo grupo lábil\(X\),, pero difiriendo sólo en la estructura de\(R\), sus tasas relativas de ionización corresponderán a las estabilidades relativas de\(R^\oplus\). Cuanto menor sea la energía de\(R^\oplus\), más rápida será la tasa de ionización. Por lo tanto, los resultados experimentales sugieren que la secuencia de estabilidades de carbocationes es\(R^\oplus\)\(\gg\) primaria\(R^\oplus\)\(\gg\) secundaria terciaria\(R^\oplus\).
Sólo por qué se observa esta secuencia es una pregunta más difícil de responder. Observe en la siguiente secuencia de estabilidad que los cationes alquilo son más estables cuanto más grupos alquilo haya en el carbono positivo:
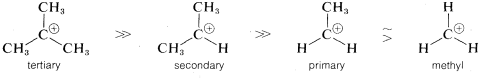
La explicación más simple de por qué esto es así es que los grupos alquilo son más polarizables que los hidrógenos. En este caso, más polarizable significa que los electrones de los grupos alquilo tienden a moverse más fácilmente hacia el carbono positivo que los de los hidrógenos. Dichos movimientos de electrones transfieren parte de la carga sobre el carbono catiónico a los grupos alquilo, extendiendo así la carga sobre un volumen mayor. Esto constituye la deslocalización de electrones, lo que da como resultado una mayor estabilidad (ver Sección 6-5A).
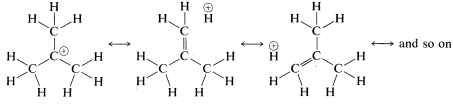
Una forma alternativa de explicar cómo se extiende la carga catiónica sobre los grupos alquilo de un catión terciario, tal como el catión terc- butilo, es escribir el catión como un híbrido de las siguientes estructuras:
Otros compuestos organohalógenos además de los compuestos de alquilo secundario y terciario pueden reaccionar por\(\text{S}_\text{N}1\) mecanismos siempre que tengan la capacidad de formar cationes de carbono razonablemente estabilizados. Los ejemplos incluyen compuestos 2-propenilo (alílico) y fenilmetil (bencílico), que al ionizar dan cationes que tienen electrones deslocalizados (ver Sección 6-6):
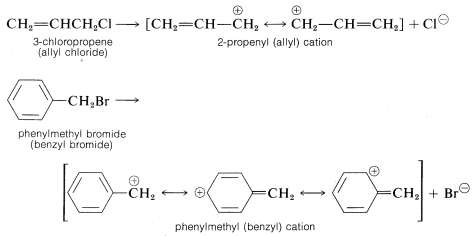
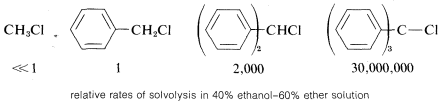
En general, cuanto más estabilizado sea el catión de carbono a partir de un haluro de alquilo, más reactivo será el compuesto en las reacciones de\(\text{S}_\text{N}1\) tipo. Esto es especialmente evidente en las reactividades de compuestos con grupos fenilo en el carbono de reacción. A medida que el número de grupos fenilo aumenta de cero a tres, la\(\text{S}_\text{N}1\) reactividad de los cloruros aumenta en más que\(10^7\) debido al aumento de la estabilización del catión de carbono por los grupos fenilo:
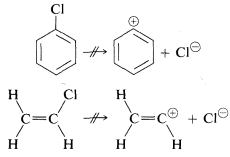
En contraste, compuestos como el clorobenceno y el cloroeteno, en los que el halógeno está unido directamente a un átomo de carbono de enlace múltiple, no presentan reacciones de\(\text{S}_\text{N}1\) tipo. Evidentemente, los cationes de carbono insaturados tales como fenilo o etenilo son apreciablemente menos estables (más difíciles de formar) que los cationes terc - alquilo:
Las razones para ello se considerarán en la Sección 14-4B.
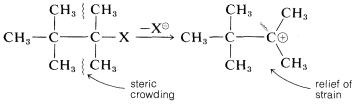
El impedimento estérico es relativamente poco importante en\(\text{S}_\text{N}1\) las reacciones debido a que la velocidad es independiente del nucleófilo. De hecho, la aceleración estérica es posible en la solvolisis de haluros de alquilo altamente ramificados a través del alivio de la compresión estérica entre los grupos alquilo en el haluro mediante la formación de un catión plano:
Junto con el efecto que\(R\) tiene sobre la velocidad a la que un compuesto alquílico\(RX\) reacciona por un\(\text{S}_\text{N}1\) mecanismo, el grupo\(R\) también afecta la naturaleza de los productos obtenidos. Los cationes alquilo intermedios\(R^\oplus\) pueden reaccionar de diversas maneras para dar productos de sustitución, eliminación y transposición. Las vías de eliminación se discuten más a fondo comenzando en la Sección 8-8, y el reordenamiento de los cationes de carbono en la Sección 8-9B.
El grupo de salida
La reactividad de un derivado de alquilo dado\(RX\), en cualquiera\(\text{S}_\text{N}1\) o\(\text{S}_\text{N}2\) reacciones, está fuertemente influenciada por el grupo lábil,\(X\). Por lo tanto, la elección del grupo lábil es una consideración importante en cualquier síntesis que implique\(\text{S}_\text{N}\) reacciones.
A partir de la discusión anterior sobre los efectos estructurales en el\(R\) grupo sobre la\(\text{S}_\text{N}\) reactividad, particularmente en\(\text{S}_\text{N}1\) las reacciones, podríamos esperar que la estabilidad de\(:X\) como ión o molécula neutra juegue un papel importante en la determinación de lo bueno o malo\(X\) que es como un abandono grupo. La estabilidad de\(:X\) es efectivamente importante -el problema es que existen varios factores que contribuyen a la estavilidad y de ahí a la labilidad del grupo de salida.
Con el propósito de identificar inicialmente grupos salientes buenos y pobres, considere el desarrollo de una síntesis práctica de éter dietílico. Una ruta es por medio de\(\text{S}_\text{N}2\) desplazamiento usando un compuesto etílico\(CH_3CH_2X\), e ion etóxido:
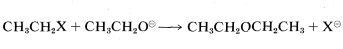
Muchos\(CH_3CH_2X\) compuestos tienen\(X\) grupos que son bastante insatisfactorios en esta reacción. Incluyen compuestos como etano, propano, etanol, etil metil éter, etilamina y etanoato de etilo; los grupos respectivos,,\(H^\ominus\),\(CH_3^\ominus\),\(HO^\ominus\),\(CH_3O^\ominus\)\(NH_2^\ominus\), y\(CH_3CO_2^\ominus\) todos pueden clasificarse como grupos salientes muy pobres. Los derivados etílicos más reactivos (ver Cuadro 8-3) incluyen los haluros, particularmente yoduro de etilo, y derivados de ácido sulfónico; los aniones correspondientes\(Cl^\ominus\),,\(Br^\ominus\)\(I^\ominus\), y\(RS \left( O_2 \right) O^\ominus\) por lo tanto son grupos salientes moderados a buenos. La Tabla 8-3 incluye datos pertinentes para las velocidades de formación de éter a partir de diversos compuestos alquílicos e ilustra que las capacidades relativas de los grupos para salir son aproximadamente las mismas en\(\text{S}_\text{N}1\) las reacciones que en\(\text{S}_\text{N}2\) las reacciones.
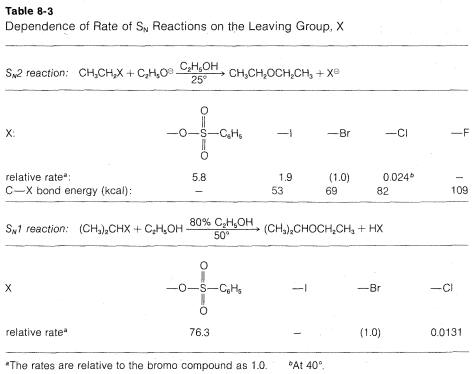
¿Por qué son grupos como\(I^\ominus\) y\(RSO_3^\ominus\) buenos grupos de salida, mientras que otros como\(H^\ominus\),\(HO^\ominus\) y\(NH_2^\ominus\) son pobres? La correlación más simple es con la fuerza de\(HX\) como un ácido. Esto es muy razonable porque la facilidad de pérdida de\(X^\ominus\), como de\(\left( CH_3 \right) C-X\) en una\(\text{S}_\text{N}1\) reacción, se esperaría que estuviera relacionada, en cierta medida al menos, con la facilidad de ionización de\(H-X\) a\(H^\oplus\) y\(X^\ominus\). Por lo tanto cuanto más fuerte\(HX\) sea como ácido, mejor\(X\) será como grupo de salida. Así\(HF\) es un ácido relativamente débil y no\(F^\ominus\) es un grupo lábil muy bueno;\(H-I\) es un ácido muy fuerte y\(I^\ominus\) es un buen grupo lábil. El orden habitual de reactividad de los haluros de alquilo,\(R-I\)\(>\)\(R-Br\)\(>\)\(R-Cl\)\(>\)\(R-F\) (cuando\(R\) es el mismo grupo en todas partes), está de acuerdo con las fuerzas ácidas de los ácidos halógenos. De igual manera,\(CF_3CO_2-\) es un grupo saliente mucho mejor que\(CH_3CO_2-\), y encontramos que el ácido trifluoroetanoico,\(CF_3CO_2H\) es un ácido varios miles de veces más fuerte que el ácido etanoico,\(CH_3CO_2H\). Por la misma razón,\(CF_3SO_3^\ominus\) es un mejor grupo de salida que\(CH_3XO_3^\ominus\).
Esta correlación se puede extender fácilmente a grupos que dejan como neutrales\(X:\). Por ejemplo,\(ROH_2^\oplus \rightarrow R^\oplus + H_2O\) ocurre mucho más fácilmente que\(ROH \rightarrow R^\oplus + OH^\ominus\) y sabemos que\(H_3O^\oplus\) es un ácido más fuerte (o mejor donante de protones) que\(H_2O\).
La relación entre\(X^\ominus\) como grupo lábil y\(HX\) como ácido es muy útil porque hay mucha información disponible sobre las fuerzas ácidas. Sin embargo, no es una explicación muy fundamental a menos que podamos explicar por qué algunos ácidos son ácidos fuertes y otros son ácidos débiles. Un factor es la fuerza del\(H-X\) enlace, pero aquí hay que recordar que las fuerzas de enlace habituales son para la disociación a radicales o átomos, no iones, y para el gas, no para soluciones. Si escribimos los pasos que relacionan la energía de enlace y disociación con la energía de disociación iónica en solución, vemos que para variaciones en\(X\), además de la energía de enlace, la afinidad electrónica de\(X \cdot\), la energía de solvatación de\(X^\ominus\), y la energía de solvatación de\(HX\), también será factores contribuyentes.

Mejora de las habilidades de los grupos de salida por catálisis electrófila
En general, un grupo lábil que sale como molécula neutra es un grupo saliente mucho mejor que uno que sale como anión. Los alcoholes,\(ROH\), son particularmente no reactivos en\(\text{S}_\text{N}\) las reacciones debido a que\(OH^\ominus\) es un grupo saliente muy pobre. Sin embargo, si está presente un ácido fuerte, la reactividad del alcohol se mejora enormemente. El ácido funciona donando un protón al oxígeno del alcohol, transformando así la función hidroxilo en\(ROH_2^\oplus\), que tiene un grupo saliente mucho mejor,\(H_2O\), en lugar de\(OH^\ominus\). Las\(\text{S}_\text{N}\) reacciones de éteres y ésteres son catalizadas por ácidos por la misma razón.
Las sales de metales pesados, particularmente las de plata, mercurio y cobre, catalizan\(\text{S}_\text{N}1\) las reacciones de haluros de alquilo de la misma manera que los ácidos catalizan las\(\text{S}_\text{N}\) reacciones de los alcoholes. Un ion de metal pesado funciona complejando con los electrones no compartidos del haluro, haciendo así que el grupo lábil sea un haluro metálico en lugar de un ion haluro. Esta aceleración de las velocidades de las reacciones de haluro es la base para una prueba cualitativa para haluros de alquilo con nitrato de plata en solución de etanol:
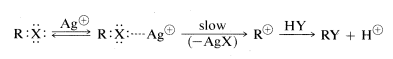
El haluro de plata precipita a una velocidad que depende de la estructura del grupo alquilo,\(>\) primario terciario\(>\) secundario. Los haluros terciarios generalmente reaccionan inmediatamente a temperatura ambiente, mientras que los haluros primarios requieren calentamiento. Que los complejos realmente se forman entre los haluros orgánicos y el ion plata se indica por un aumento en la solubilidad en agua en presencia de iones de plata para aquellos haluros que son lentos en la formación de carboncationes.
El reactivo nucleofílico
La nucleofilia de un reactivo particular (\(:Y\),\(:Y^\ominus\), o\(H \ddot{Y}\)) puede definirse como su capacidad para donar un par de electrones a otro átomo (ver Sección 8-1). De hecho, la\(\text{S}_\text{N}2\) reactividad de un reactivo hacia un derivado metílico se puede tomar para medir su nucleofilia hacia el carbono. Las velocidades de reacción relativas de algunos nucleófilos hacia el bromuro de metilo se enumeran en orden de nucleofilia creciente en la Tabla 8-4, junto con sus basicidades medidas por\(K_b\). Se pueden hacer generalizaciones importantes a partir de estos datos siempre que se reconozca que solo pueden aplicarse a disolventes hidroxílicos.
1. Para los átomos que representan cualquier grupo (columna) de la tabla periódica, la nucleofilia aumenta al aumentar el número atómico:\(I^\ominus\)\(>\)\(Br^\ominus\)\(>\)
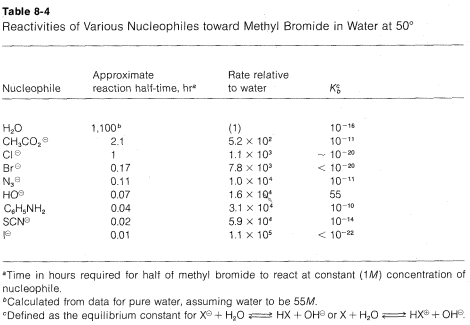
\(Cl^\ominus\)\(>\)\(F^\ominus\);\(HS^\ominus\)\(>\)\(HO^\ominus\);\(PH_3\)\(>\)\(NH_3\). Así, siendo otras cosas iguales, los átomos más grandes son mejores nucleófilos.
2. Para los nucleófilos que tienen el mismo número atómico del átomo entrante (por ejemplo, nucleófilos de oxígeno), suele existir una buena correlación entre la basicidad del reactivo y su nucleofilia. Así una base débil tal como\(CH_3CO_2^\ominus\) es un nucleófilo más pobre que una base fuerte como\(^\ominus OH\). Cuanto más pobre\(X^\ominus\) es como grupo de salida, mejor es como grupo entrante.
3. Para nucleófilos de diferentes números atómicos, la nucleofilia generalmente no es basicidad paralela. Por ejemplo, para los halógenos la secuencia de reactividad\(I^\ominus\)\(>\)\(Br^\ominus\)\(>\)\(Cl^\ominus\) es opuesta a la secuencia de basicidad\(Cl^\ominus\)\(>\)\(Br^\ominus\)\(>\)\(I^\ominus\). De igual manera, aniones de azufre como\(HS^\ominus\) son mejores nucleófilos pero bases más débiles que los oxianiones correspondientes como\(HO^\ominus\).
4. Una serie de agentes nucleofílicos, que son muy reactivos en\(\text{S}_\text{N}2\) las reacciones, son del tipo\(X-Y\), donde ambos átomos tienen pares de electrones no compartidos. Los ejemplos incluyen\(HOO^\ominus\)\(H_2NO^\ominus\),\(ClO^\ominus\), y\(H_2NNH_2\), todos los cuales son más reactivos que los nucleófilos estrechamente relacionados\(HO^\ominus\) y\(NH_3\).
¿Por qué la correlación entre basicidad y nucleofilcidad es tan pobre para átomos de diferente número atómico? ahora queda claro a partir de muchas investigaciones que el efecto dominante está asociado con diferencias en las energías de solvatación de los iones, tal como se define para los iones haluro mediante las siguientes ecuaciones:
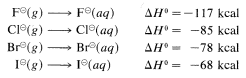
Las energías de solvatación de los iones pequeños con carga concentrada siempre son mayores que las de los iones grandes con carga difusa.
Cuando un ion participa en un ataque nucleofílico sobre carbono, debe desprenderse de algunas de las moléculas de disolvente que lo estabilizan en solución. De lo contrario, el ion no puede acercarse lo suficiente al carbono, al que se adherirá, para comenzar a formar un enlace. El desprendimiento de moléculas de disolvente será menos favorable para un ion pequeño que para un ion grande. En consecuencia, esperamos\(Cl^\ominus\) ser menos reactivos que\(I^\ominus\).
Una fuerte evidencia de efectos de solvatación sobre la reactividad es proporcionada por el hecho de que el ion cloruro es más reactivo que el yoduro en disolventes que tienen bajas energías de solvatación para aniones (ver Sección 8-7F). Además, en la fase gaseosa donde los efectos de solvatación están ausentes,\(F^\ominus\) es más reactivo que cualquiera de los otros iones haluro hacia el clorometano:
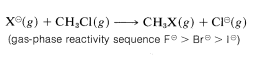
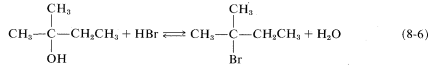
Se debe reconocer que\(\text{S}_\text{N}\) las reacciones pueden ser reversibles cuando tanto el grupo\(X\) de salida como el grupo entrante\(Y\) son buenos grupos de entrada y salida, respectivamente. En tales circunstancias, la posición del equilibrio a menudo se puede cambiar ajustando adecuadamente las condiciones de reacción. Así, el bromuro de hidrógeno\(48\%\) acuoso puede convertir alcoholes en bromidos de alquilo (Ecuación 8-6, dirección hacia adelante), mientras que la reacción inversa (hidrólisis) se logra mediante una alta concentración de agua:
La naturaleza del solvente
Las velocidades de\(\text{S}_\text{N}\) reacción son sensibles a la naturaleza y composición del disolvente. Esto es fácil de entender para\(\text{S}_\text{N}1\) las reacciones porque el poder ionizante de un disolvente es crucial para la facilidad de formación de iones\(\overset{\oplus}{R}\) y\(\overset{\ominus}{X}\) de\(RX\).
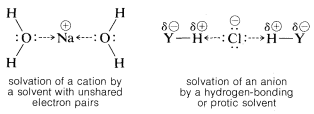
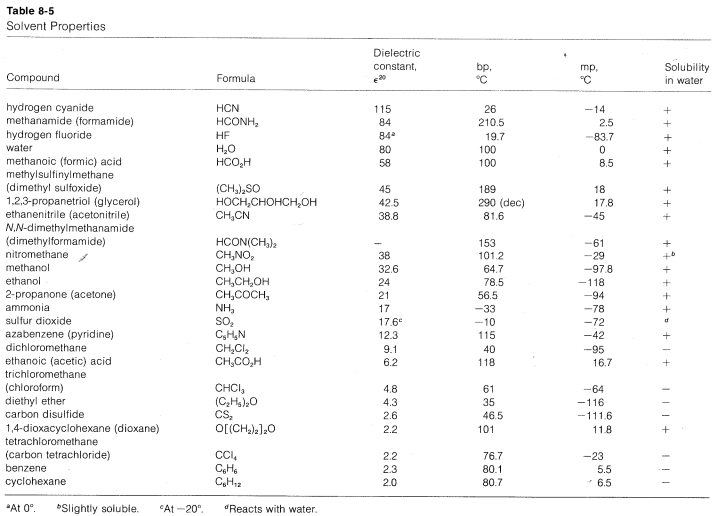
En realidad, dos factores son relevantes en cuanto a la capacidad ionizante de los solventes. Primero, una constante dieléctrica alta aumenta la potencia ionizante al facilitar la separación de iones. Esto se debe a que la fuerza entre las partículas cargadas varía inversamente con la constante dieléctrica del medio. \(^7\)Así, el agua, con una constante dieléctrica de 80, es 40 veces más efectiva que un hidrocarburo con una constante dieléctrica de 2. Segundo, y generalmente más importante, es la capacidad del disolvente para solvatar los iones separados. Los cationes son solvatados de manera más efectiva por compuestos de elementos en la primera fila de la tabla periódica que tienen pares de electrones no compartidos. Ejemplos son amoníaco, agua, alcoholes, ácidos carboxílicos, dióxido de azufre y metilsulfinilmetano [dimetilsulfóxido,\(\left( CH_3 \right)_2 SO\)]. Las anoínas son solvatadas más eficientemente por disolventes que tienen hidrógeno unido a un elemento fuertemente elecronegativo\(Y\) por lo que el\(H-Y\) enlace está fuertemente polarizado como\(\overset{\delta \oplus}{H} \: \cdot \: \cdot \: \cdot \: \overset{\delta \ominus}{Y}\). Tales solventes generalmente se llaman solventes próticos. Los solventes próticos forman enlaces de hidrógeno con el grupo lábil, que ayudan a la ionización de la misma manera que el ion plata cataliza la ionización de haluros de alquilo (Sección 8-7D). Podemos representar la solvatación por las siguientes fórmulas estructurales, pero hay que reconocer que el número de moléculas de disolvente involucradas en interaciton cercanas puede ser tan grande como cuatro o seis, o tan pequeño como uno:
Los solventes ionizantes más efectivos son aquellos que solvatan eficazmente tanto aniones como cationes. El agua alcanza un excelente compromiso con respecto a las características estructurales que componen el poder ionizante, es decir, la constante dieléctrica y la capacidad de solvatación. De esto, se espera que el cloruro de terc- butilo se ionice mucho más fácilmente en agua que en éter, ya que los éteres pueden solvatar solo cationes de manera efectiva, mientras que el agua puede solvatar tanto aniones como cationes. El hecho es que\(\text{S}_\text{N}1\) las ionizaciones suelen ser tan difíciles que rara vez ocurren\(\text{S}_\text{N}1\) reacciones en disolventes que no pueden solvatar eficazmente tanto aniones como cationes, aunque la constante dieléctrica del disolvente sea alta. La solvatación por enlaces de hidrógeno es especialmente útil para ayudar a la ionización. Los disolventes que no pueden proporcionar tales enlaces de hidrógeno [por ejemplo\(CH_3OCH_3\),\(\left( CH_3 \right)_3 N\),\(CH_3NO_2\),\(CH_3CN\),,\(\left( CH_3 \right)_2 SO\)] generalmente son pobres para\(\text{S}_\text{N}1\) las reacciones. Estos disolventes se denominan disolventes apróticos. Una excepción importante es el dióxido de azufre líquido\(SO_2\), que promueve\(\text{S}_\text{N}1\) la ionización al tener una constante dieléctrica alta y poder solvatar tanto aniones como cationes.
En el Cuadro 8-5 se presenta una lista de solventes próticos y apróticos, sus constantes dieléctricas, puntos de ebullición y puntos de fusión. Esta tabla será útil en la selección de disolventes para reacciones de sustitución nucleofílica.
Con respecto a\(\text{S}_\text{N}2\) las reacciones, el disolvente puede afectar profundamente la reactividad de un nucleófilo dado. Así, aniones como\(Cl^\ominus\) y\(CN^\ominus\), que son débilmente nucleófilos en disolventes hidroxílicos y en disolventes ionizantes pobres como la 2-propanona (acetona), se vuelven muy significativamente nucleófilos en sovlentes apróticos polares como\(\left( CH_3 \right)_2 SO\). La razón es que para sales como\(NaCl\) y\(NaCN\) el disolvente aprótico preferentemente solvata el catión, dejando el anión relativamente desnudo. Esta disociación del anión del catión junto con su pobre solvatación hace que el anión sea anormalmente reactivo como nucleófilo.
\(^6\)Las letras griegas\(\alpha\),\(\beta\),\(\gamma\) se utilizan aquí no como nomenclatura, sino para designar las posiciones a lo largo de una cadena carbonada a partir de un grupo funcional,\(X\):\(C_\omega \cdots C_\delta-C_\gamma-C_\beta-C_\alpha-X\) (véase también la Sección 7-10).
\(^7\)Específicamente, la fuerza electrostática\(= q_1 q_2/r_{12}^2 \epsilon\) en la que\(q_1\) y\(q_2\) están las cargas,\(r_{12}\) es la distancia entre las cargas, y\(\epsilon\) es la constante dieléctrica del medio;\(\epsilon = 1\) para un vacío.
Colaboradores y Atribuciones
- John D. Robert and Marjorie C. Caserio (1977) Basic Principles of Organic Chemistry, second edition. W. A. Benjamin, Inc. , Menlo Park, CA. ISBN 0-8053-8329-8. This content is copyrighted under the following conditions, "You are granted permission for individual, educational, research and non-commercial reproduction, distribution, display and performance of this work in any format."