
28.2: Absorción de luz, fluorescencia y fosforescencia
- Page ID
- 72477

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)La radiación electromagnética en la región ultravioleta y visible abarca un rango de longitud de onda de aproximadamente\(800\) -\(100 \: \text{nm}\) correspondiente a las energías de\(36\) -\(286 \: \text{kcal mol}^{-1}\). La absorción de dicha radiación por las moléculas no debe considerarse equivalente a la simple excitación por energía térmica de\(36\) -\(286 \: \text{kcal mol}^{-1}\). En cambio, toda la energía del cuántico de luz es absorbida en la excitación de un electrón a un orbital de alta energía, generalmente antiunión, (Sección 9-9). Un punto importante de tales procesos es que ocurren más rápidamente de lo que los átomos vibran en los enlaces (principio Franck-Condon). El corto tiempo de transición de un electrón entre los estados tierra y excitado está en completo contraste con lo que ocurre durante la absorción de un cuántico de energía de radiofrecuencia en espectroscopía de RMN, en donde el proceso de absorción puede ser lento en comparación con las reacciones químicas (Sección 27-1). Por lo tanto, una molécula excitada electrónicamente es, en el primer instante en que se produce\(\left( <10^{-13} \: \text{sec} \right)\), al igual que la molécula de estado fundamental en cuanto a posiciones y energías cinéticas de los átomos van, pero tiene una configuración electrónica muy diferente. Lo que sucede en este punto depende de varios factores, algunos de los cuales se pueden ilustrar mejor mediante diagramas de energía del tipo utilizado anteriormente (Sección 21-1). Consideraremos moléculas diatómicas, pero el argumento puede extenderse a sistemas más complicados.
Considere la Figura 28-1, que muestra curvas esquemáticas de potencial-energía para una molécula\(\ce{A-B}\) en estado fundamental\(\left( \ce{A-B} \right)\) y en estados electrónicos excitados\(\left( \ce{A-B} \right)^*\). Hemos señalado anteriormente (Sección 6-1) que en los estados básicos de la mayoría de las moléculas todos los electrones están emparejados; los estados excitados también pueden tener todos los electrones emparejados. Tales estados con electrones emparejados se denominan estados singlete. Pero, debido a que la unión es más débil en los estados excitados, la longitud promedio del enlace\(r_e\) entre los núcleos es mayor en el estado excitado que en el estado fundamental. Por esta razón, la curva superior\(\left( S_1 \right)\) de la Figura 28-1 se desplaza hacia una longitud de enlace promedio mayor en relación con la curva inferior o de estado fundamental\(\left( S_0 \right)\).
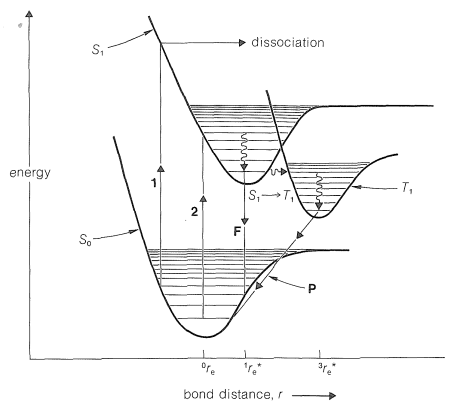
Los estados excitados también pueden tener electrones desapareados. Los estados con dos electrones desapareados se denominan estados tripletes\(\left( T \right)\) y normalmente son más estables que los estados singlete correspondientes porque, por regla de Hund, se espera menos repulsión interelectrónica con electrones desapareados que emparejados (Secciones 6-1 y 21-9A). (Para mayor claridad, a la curva potencial-energía para el estado triplete excitado\(\left( T_1 \right)\) de\(\ce{A-B}\) se le da una distancia de enlace de equilibrio irrealistamente larga, lo que la coloca a la derecha de la curva para el\(S_1\) estado en la Figura 28-1). Las configuraciones electrónicas para los\(\left( T_1 \right)\) estados singlete\(\left( S_0 \right)\) de tierra\(\left( S_1 \right)\), singlete excitado y triplete de los\(\sigma\) electrones de una molécula diatómica se muestran en la Figura 28-2. Este diagrama será útil para interpretar las transiciones entre\(S_0\),\(S_1\), y\(T_1\) estados que se muestran en la Figura 28-1, y que ahora discutiremos con más detalle.

Cuando una molécula absorbe suficiente energía radiante para provocar excitación electrónica, el giro del electrón excitado permanece sin cambios en la transición. Es decir, las moléculas de estado fundamental con electrones emparejados\(\left( S_0 \right)\) dan estados excitados con electrones emparejados\(\left( S_1 \right)\), no estados tripletes\(\left( T_1 \right)\). La transición marcada\(1\) en la Figura 28-1 corresponde a una\(\left( S_0 \rightarrow S_1 \right)\) transición singlet-singlete desde un nivel vibracional relativamente alto de\(\ce{A-B}\). El cambio de energía ocurre sin cambio en\(r\) (principio Franck-Condon), y la energía electrónica de la\(\ce{A-B}^*\) molécula así producida se ve que está por encima del nivel requerido para la disociación de\(\ce{A-B}^*\). La vibración de la molécula excitada, por lo tanto, no tiene fuerza restauradora y conduce a la disociación\(\ce{A}\) y los\(\ce{B}\) átomos. En contraste, la transición marcada\(2\) conduce a un estado vibracional excitado de\(\ce{A-B}^*\), que no se espera que se disocie sino que puede perder energía vibratoria al entorno y bajar a un estado vibracional inferior. Esto se llama “relajación vibracional” y por lo general requiere aproximadamente\(10^{-12} \: \text{sec}\). El estado excitado vibracionalmente “relajado” puede regresar al estado fundamental con emisión de radiación (transición\(F\),\(S_1 \rightarrow S_0\)); esto se conoce como fluorescencia, siendo la longitud de onda de fluorescencia diferente a la de la luz original absorbida. Normalmente, la fluorescencia, si ocurre, ocurre en\(10^{-9}\) hasta\(10^{-7} \: \text{sec}\) después de la absorción de la radiación original.
En muchos casos, el estado excitado\(\left( S_1 \right)\) puede regresar al estado fundamental\(\left( S_0 \right)\) por procesos no radiativos. Los procesos más importantes son:
1. Por reacción química, a menudo con moléculas circundantes. Este proceso forma la base de gran parte de la fotoquímica orgánica, la cual será descrita en una sección posterior.
2. Por transferencia de su exceso de energía electrónica a otras moléculas. Este tipo de transferencia de energía también es un aspecto muy importante de la fotoquímica, y volveremos a ella en breve.
3. Por decaimiento a través de un estado de menor energía. Si, por ejemplo, las curvas de potencial-energía para los estados singlete superior e inferior estuvieran más juntas que las mostradas en la Figura 28-1, en realidad pueden cruzarse en algún punto, proporcionando así una vía\(S_1\) para relajarse\(S_0\) sin fluorescencia. Pero ¿qué pasa con la decadencia\(S_1\) a través del estado triplete\(\left( T_1 \right)\)?
La conversión de un estado excitado singlete a un estado triplete\(\left( S_1 \rightarrow T_1 \right)\) es energéticamente favorable pero generalmente ocurre bastante lentamente, de acuerdo con las reglas de selección espectroscópica, que predicen que los cambios espontáneos de espín electrónico deben tener probabilidades muy bajas. Sin embargo, si el estado singlete es suficientemente largo, el cambio de singlete y triplete\(S_1 \rightarrow T_1\), (a menudo llamado cruce intersistema) puede ocurrir para una proporción muy considerable de las moléculas singlete excitadas.
El estado triplete, al igual que el estado singlete, puede regresar al estado fundamental por procesos no radiativos, pero en muchos casos\(\left( T_1 \rightarrow S_0 \right)\) se produce una transición radiativa, aunque tiene baja probabilidad. Tales transiciones dan como resultado la emisión de luz de longitud de onda considerablemente más larga que la absorbida originalmente o que resulta de la fluorescencia. Este tipo de transición radiativa se denomina fosforescencia (transición\(P\) en la Figura 28-1). Debido a que la fosforescencia es un proceso con una probabilidad baja, el\(T_1\) estado puede persistir de fracciones de segundo a muchos segundos. Para el benceno en\(-200^\text{o}\), la absorción de luz\(254 \: \text{nm}\) conduce a fluorescencia centrada en\(290 \: \text{nm}\) y fosforescencia en\(340 \: \text{nm}\). La vida media del estado triplete del benceno en\(-200^\text{o}\) es\(7 \: \text{sec}\).
El Grupo Carbonilo
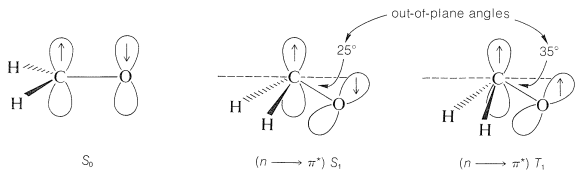
En discusiones previas sobre espectros de absorción electrónica (Sección 9-9), se han identificado dos tipos diferentes de transiciones en los espectros de compuestos carbonílicos simples como 2-propanona o metanal. Uno implica la excitación de un electrón en un\(n\) orbital no enlazante sobre oxígeno a un\(\left( \pi^* \right)\) orbital antienlace del doble enlace carbono-oxígeno (una\(n \rightarrow \pi^*\) transición), y el otro implica la excitación de un electrón en el\(\left( \pi \right)\) orbital de enlace al orbital antienlace correspondiente (a \(\pi \rightarrow \pi^*\)transición). Estos cambios se muestran para el metanal en la Figura 28-3. Además de las transiciones ya discutidas, el metanal muestra una fuerte absorción en\(175 \: \text{nm}\), que posiblemente sea\(n \rightarrow \sigma^*\), o de lo contrario\(\sigma \rightarrow \sigma^*\).

Aunque las\(\pi \rightarrow \pi^*\) transiciones\(n \rightarrow \pi^*\) y de la Figura 28-3 son transiciones singlete-singlete, cada uno de los dos estados excitados singlete producidos tiene un estado triplete correspondiente. En consecuencia, hay cuatro estados excitados fácilmente accesibles de un grupo carbonilo: el\(n \rightarrow \pi^*\) singlete\(\left( S_1 \right)\), el\(n \rightarrow \pi^*\) triplete\(\left( T_1 \right)\), el\(\pi \rightarrow \pi^*\) singlete\(\left( S_2 \right)\) y el\(\pi \rightarrow \pi^*\) triplete\(\left( T_2 \right)\). Las energías de estos estados electrónicos para metanal disminuyen en el orden\(S_2 > T_2 > S_1 > T_1\), aunque este orden puede no sostenerse para todos los compuestos carbonílicos.
Como veremos, los estados\(n \rightarrow \pi^*\) singlete y triplete de los compuestos carbonílicos juegan un papel importante en la fotoquímica. Los aldehídos y cetonas presentan todas las características de absorción, fluorescencia, fosforescencia y cruce entre sistemas\(\left( S_1 \rightarrow T_1 \right)\) ilustradas en la Figura 28-1. Generalmente, son más eficientes en el cruce intersistémico que los hidrocarburos insaturados, quizás porque las energías de\(S\) los\(T\) estados involucrados no son muy diferentes.
Además de que las longitudes de enlace son más largas en los estados excitados de las moléculas, las formas moleculares difieren de las de los estados básicos. Aunque el principio Franck-Condon requiere que la absorción produzca estados excitados con la misma geometría que los estados básicos, las moléculas excitadas a partir de entonces pueden relajarse a formas más estables, que pueden ser no planares y retorcidas alrededor de\(\pi\) los enlaces anteriores. El metanal es plano con una longitud de\(\ce{C-O}\) enlace de\(1.21 \: \text{Å}\) en el estado fundamental, pero en el\(\left( S_1 \right)\) estado\(n \rightarrow \pi^*\) singlete, el metanal es piramidal, con una longitud de\(\ce{C-O}\) enlace de\(1.32 \: \text{Å}\). El metanal está aún más distorsionado en el estado\(n \rightarrow \pi^*\) triplete, aunque la longitud del enlace sigue siendo aproximadamente la misma en\(1.32 \: \text{Å}\).
Transferencia de energía de excitación electrónica indirecta
Es posible producir estados electrónicos excitados de moléculas indirectamente por medio de la transferencia de energía de otras moléculas excitadas. Un ejemplo es proporcionado por la excitación de naftaleno como resultado de la transferencia de energía de la benzofenona excitada. Benzofenona,\(\ce{C_6H_5COC_6H_5}\), absorbe la luz ultravioleta con\(\lambda_\text{max} = 330 \: \text{nm}\) en una\(n \rightarrow \pi^*\) transición. La naftalina no absorbe apreciablemente en esta región. Sin embargo, la irradiación de una mezcla de benzofenona y naftaleno con\(330\)\(\text{nm}\) luz produce emisión fosforescente de naftaleno. Así, la benzofenona absorbe la luz y transfiere su exceso de energía a la naftalina, que regresa al estado fundamental por emisión. Debido a que la emisión es del estado triplete de naftaleno, la benzofenona debe estar involucrada en la excitación del naftaleno al estado triplete. Podemos escribir el proceso de la siguiente manera:

La transferencia de energía no implica un cambio neto en el espín electrónico. Para que esto se mantenga para la excitación de naftaleno de\(S_0\) a\(T_1\), la transferencia de energía debe provenir de triplete (no singlete) benzofenona). El proceso de producir estados excitados de esta manera se denomina fotosensibilización. Singlet-singlete, así como triplete-triplete, son posibles transferencias de energía, pero en todos los casos no hay cambio neto en el giro. La transferencia eficiente de energía solo será posible si\(\Delta G^0\) para la transferencia es pequeña o negativa.
Colaboradores y Atribuciones
- John D. Robert and Marjorie C. Caserio (1977) Basic Principles of Organic Chemistry, second edition. W. A. Benjamin, Inc. , Menlo Park, CA. ISBN 0-8053-8329-8. This content is copyrighted under the following conditions, "You are granted permission for individual, educational, research and non-commercial reproduction, distribution, display and performance of this work in any format."