
28.3: Fotoquímica orgánica
- Page ID
- 72491

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Se sabe que una extraordinaria variedad de reacciones de compuestos orgánicos ocurren bajo la influencia de la luz visible y ultravioleta. Algunos de estos, como la halogenación fotoquímica de alcanos y la fotosíntesis en plantas verdes, ya han sido discutidos (ver Secciones 4-4D y 20-9). No es nuestro propósito aquí revisar en detalle la fotoquímica orgánica, sino que mencionaremos algunos tipos de reacciones fotoquímicas importantes y mostraremos cómo estas pueden ser explicadas por los principios discutidos en la sección anterior.
Los compuestos tienen un comportamiento químico muy diferente en sus estados excitados en comparación con sus estados básicos. No sólo la energía es mucho mayor, sino que la geometría molecular y las configuraciones electrónicas son diferentes. Intuitivamente, esperamos que los estados excitados de las moléculas, en los que dos electrones ocupan orbitales separados sin llenar, tengan un carácter dirradical sustancial. Este es el caso, sobre todo para los estados tripletes, como veremos.
Reacciones de fotodisociación
Hemos mencionado cómo las moléculas de cloro se disocian a átomos de cloro al absorber la luz casi ultravioleta y con ello provocan la cloración de la cadena radical de hidrocarburos saturados (Sección 4-4D). La cloración fotoquímica es un ejemplo de una reacción fotoquímica que puede tener un alto rendimiento cuántico, es decir, se pueden generar muchas moléculas de producto de cloración por cuántica de luz absorbida. Se dice que el rendimiento cuántico de una reacción es unidad cuando\(1 \: \text{mol}\) el reactivo se convierte en producto (s) por einstein\(^1\) de luz absorbida. El símbolo para el rendimiento cuántico suele ser\(\Phi\).
El vapor de 2-propanona (acetona) se somete a una reacción de fotodisociación con\(313\) -\(\text{nm}\) luz con\(\Phi\) algo menos que la unidad. La absorción de luz por 2-propanona da como resultado la formación de un estado excitado que tiene suficiente energía para sufrir la escisión de un\(\ce{C-C}\) enlace (el enlace más débil de la molécula) y formar un radical metilo y un radical etanoilo. Esta es una reacción fotoquímica primaria:
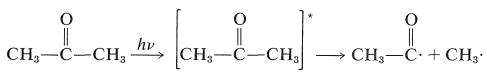 \(\tag{28-1}\)
\(\tag{28-1}\)
Los pasos posteriores son reacciones oscuras.
A temperaturas muy superiores a la temperatura ambiente, el radical etanoilo se descompone para dar otro radical metilo y monóxido de carbono:
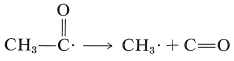 \(\tag{28-2}\)
\(\tag{28-2}\)
Si esta reacción llega a completarse, los principales productos de reacción son etano y monóxido de carbono:
\[2 \ce{CH_3} \cdot \rightarrow \ce{CH_3-CH_3} \tag{28-3}\]
Si el radical etanoilo no se descompone completamente, entonces también se forma algo de 2,3-butanodiona. Esta reacción es bastante importante a temperatura ambiente o por debajo:
 \(\tag{28-4}\)
\(\tag{28-4}\)
También se forman cantidades menores de metano y cetena como resultado de reacciones de desproporción que implican transferencias de átomos de hidrógeno de los tipos que hemos encontrado anteriormente en reacciones radicales (ver Sección 10-8C):
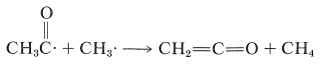 \(\tag{28-5}\)
\(\tag{28-5}\)
Las reacciones formadoras de producto, Ecuaciones 28-2 a 28-5, dependen todas del evento fotoquímico primario, Ecuación 28-1, que rompe el\(\ce{C-C}\) enlace con el grupo carbonilo. Esta escisión se ha denominado un proceso Norrish tipo I después del eminente fotoquímico, R. G. W. Norrish:\(^2\)
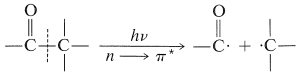
Otra reacción fotoquímica es importante para las cetonas que tienen al menos un\(\gamma\) hidrógeno en una cadena conectada al grupo carbonilo, como en
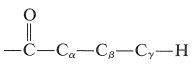
En esta vía (proceso Norrish tipo II), la escisión se produce en el\(\ce{C}_\alpha \ce{-C}_\beta\) enlace para dar, como producto principal, una cetona de longitud de cadena más corta y un alqueno. Así, para 2-pentanona:
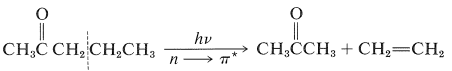
Esta reacción ocurre de una manera interesante. Cualquiera que sea la naturaleza del estado entonces\(n \rightarrow \pi^*\) excitado\(T_1\),\(S_1\) o, la reacción fotoquímica primaria es la abstracción de un átomo de hidrógeno del\(\gamma\) carbono por el oxígeno carbonilo para dar el dirradical,\(1\):
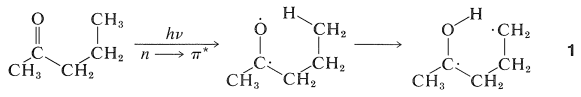
Las reacciones oscuras posteriores se entienden fácilmente como típicas de dirradicales. La escisión de\(1\) at\(\ce{C}_\alpha \ce{-C}_\beta\) da eteno y un enol, que se reorganiza a la cetona. Alternativamente,\(1\) se puede ciclar a un ciclobutanol:
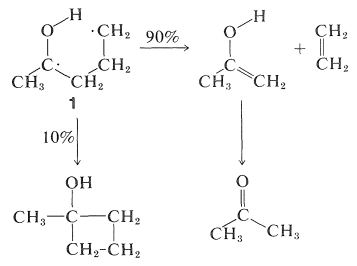
Se ha encontrado una variedad de reacciones de fotodisociación con cetonas, pero los productos casi siempre se pueden explicar como resultado de la escisión Norrish tipo I y/o II. Ejemplos son:
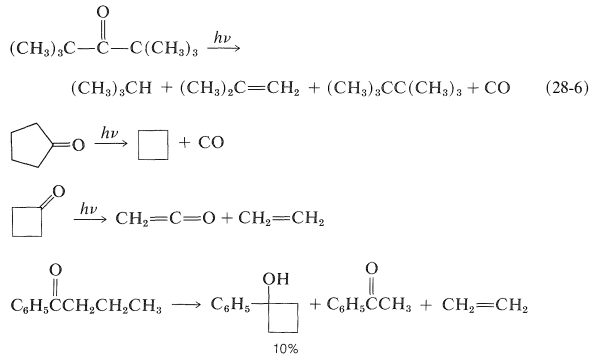
Fotorreducción de Diaril Cetonas
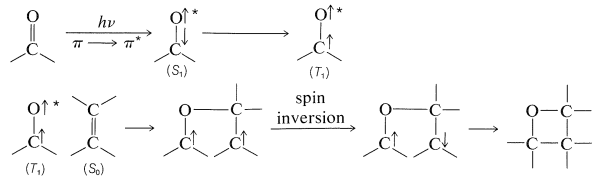
Las diaril cetonas no experimentan fotodisociación de la misma manera que las alquilcetonas, probablemente debido a que la escisión a fenilo y otros radicales arilo es desfavorable (Cuadro 4-6). Sin embargo, las cetonas aromáticas son fotoquímicamente reactivas en presencia de compuestos que pueden donar un átomo de hidrógeno, con el resultado de que el grupo carbonilo se reduce. En efecto, una de las reacciones fotoquímicas clásicas de la química orgánica es la formación de 1,1,2,2-tetrapenil-1,2-etandiol (\(3\), benzopinacol) por la acción de la luz sobre una solución de difenilmetanona (\(2\), benzofenona) en alcohol isopropílico. El rendimiento es cuantitativo.
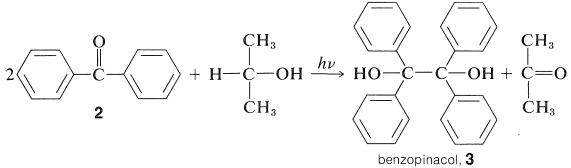
La luz es absorbida por\(2\) y la cetona activada resultante,\(2^*\), elimina un hidrógeno del alcohol isopropílico:
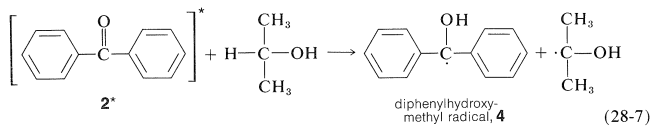
El benzopinacol resulta de la dimerización de los radicales,\(4\):
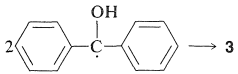
Dado que los rendimientos cuánticos de 2-propanona y benzopinacol son ambos casi la unidad cuando la intensidad de la luz no es alta, es evidente que dos de los radicales\(4\),, deben formarse por cada molécula de\(2\) que se active por la luz. Esto es posible si el radical 2-hidroxi-2-propilo formado por la Ecuación 28-7 reacciona\(2\) para dar un segundo radical difenilhidroximetilo:
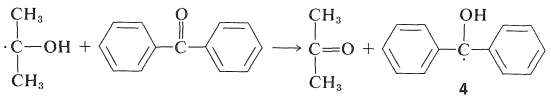
Esta reacción es energéticamente favorable debido a la mayor posibilidad de deslocalización del electrón impar en\(4\) que en el radical 2-hidroxi-2-propilo.
La formación fotoquímica de\(3\) también se puede lograr a partir de difenilmetanona\(2\), y difenilmetanol,\(5\):

El mecanismo es similar al del alcohol isopropílico como agente reductor:
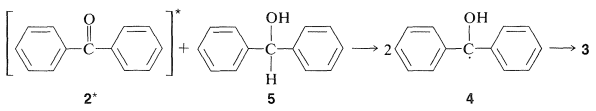
Se cree que esta reducción implica el estado triplete de\(2\) por el siguiente argumento: La formación de\(3\) es razonablemente eficiente incluso cuando la concentración del alcohol,\(5\) es baja; por lo tanto, cualquiera que sea el estado excitado de la cetona,\(2^*\), que acepta un átomo de hidrógeno de \(5\), debe ser uno bastante longevo. Debido a que las soluciones de no\(2\) muestran fluorescencia visible, deben convertirse rápidamente a otro estado de vida más larga que el singlete\(\left( S_1 \right)\). El estado de larga vida es entonces lo más razonablemente un estado triplete. De hecho, si se agrega naftaleno a la mezcla de reacción, la formación de benzopinacol,\(3\) se inhibe drásticamente debido a que el triplete de benzofenona transfiere energía al naftaleno más rápidamente de lo que reacciona con el alcohol,\(5\) (ver Sección 28-1A).
Isomerización Fotoquímica de Alquenos Cis y Trans
Un problema importante en muchas síntesis es producir el isómero deseado de un par cis-trans de alquenos. El problema no surgiría si fuera posible isomerizar el isómero no deseado al isómero deseado. En muchos casos tales isomerizaciones se pueden llevar a cabo fotoquímicamente. Un ejemplo típico es proporcionado por cis- y trans-1,2-difenileteno (estilbeno):
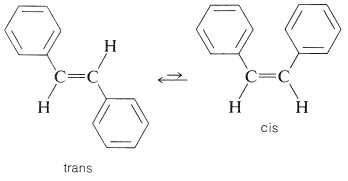
Aquí la forma trans está fácilmente disponible por una variedad de reacciones y es más estable que el isómero cis porque tiene menos impedimento estérico. Sin embargo, es posible producir una mezcla que contiene principalmente isómero cis irradiando una solución del isómero trans en presencia de un fotosensibilizador adecuado. Este proceso de ninguna manera contraviene las leyes de la termodinámica porque la entrada de energía radiante permite que el punto de equilibrio sea desplazado de lo que sería normalmente.
La isomerización parece ocurrir por la siguiente secuencia: El sensibilizador, generalmente una cetona como la benzofenona o la 1- (2-naftil) etanona, se eleva por una\(n \rightarrow \pi^*\) transición del estado fundamental singlete\(\left( S_0 \right)\) a un estado excitado\(\left( S_1 \right)\) por absorción de luz. El cruce entre sistemas se produce entonces rápidamente para dar el estado triplete\(\left( T_1 \right)\) del sensibilizador:

El siguiente paso es la excitación del alqueno por transferencia de energía desde el estado triplete del sensibilizador. Recuerde, el espín electrónico neto se conserva durante la transferencia de energía, lo que significa que el alqueno se excitará al estado triplete:
\[^3\text{Sens}^* \left( \uparrow \uparrow \right) + ^1\text{Alkene} \left( \uparrow \downarrow \right) \rightarrow ^1\text{Sens} \left( \uparrow \downarrow \right) + ^3\text{Alkene}^* \left( \uparrow \uparrow \right)\]
El estado triplete del alqueno es más estable cuando los\(p\) orbitales, que conforman el\(\pi\) sistema normal del doble enlace, no son paralelos entre sí (Figura 6-17). Por lo tanto, si el proceso de transferencia de energía conduce inicialmente a un triplete plano, éste se convierte rápidamente a la forma no planar más estable. La excitación del isómero cis o trans del alqueno parece conducir a un estado triplete común, como se muestra en la Figura 28-4.
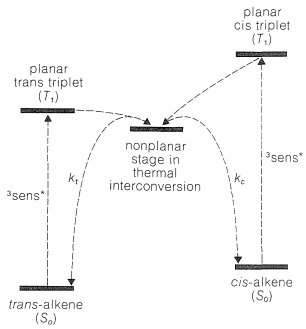
El paso final en la isomerización es la descomposición del triplete alqueno al estado fundamental. Esto ocurre ya sea por emisión de luz (fosforescencia) o por tener la energía triplete convertida en energía térmica sin emisión de luz. De cualquier manera, se podría formar el isómero cis o trans y, como puede verse en la Figura 28-4, la relación de isómeros producidos depende de las tasas relativas de descomposición del triplete de alqueno a los isómeros del estado fundamental,\(k_c/k_t\). Esta relación resulta favorecer la formación del isómero menos estable. Por lo tanto, siempre que ambos isómeros puedan ser fotosensibilizados de manera eficiente, la irradiación sensibilizada de cualquiera de ellos conducirá finalmente a una mezcla de ambos, en la que predomina el isómero termoquímicamente menos estable. El sensibilizador debe tener una energía triplete superior a la energía triplete del alqueno para que se produzca la transferencia de energía, y el punto fotoestacionario o de equilibrio es independiente de la naturaleza del sensibilizador cuando este último transfiere energía de manera eficiente a ambos isómeros cis y trans. En el uso práctico del equilibrio fotoquímico sensibilizado de isómeros cis y trans, normalmente es necesario realizar experimentos piloto para determinar qué sensibilizadores son útiles.
Otro ejemplo de cómo se puede usar la isomerización fotoquímica se proporciona mediante el equilibrio de la\(Z\) forma\(E\) y de 1-bromo-2-fenil-1-propeno:
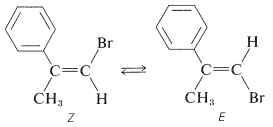
El\(E\) isómero se forma hasta el punto de\(95\%\) en la deshidrohalogenación de 1,2-dibromo-2-fenilpropano:
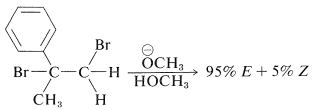
La fotoisomerización del producto de eliminación con 1- (2-naftil) etanona como sensibilizador produce una mezcla que contiene\(85\%\) del\(Z\) isómero.
Reacciones de ciclación fotoquímica
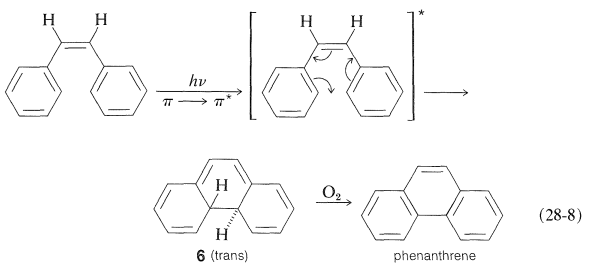
Bien se puede preguntar por qué la isomerización de alquenos discutida en la sección anterior requiere un sensibilizador. ¿Por qué no se puede lograr el mismo resultado mediante irradiación directa? Una razón es que un estado excitado\(\pi \rightarrow \pi^*\) singlete\(\left( S_1 \right)\) producido por irradiación directa de un alqueno o areno cruza al estado triplete de\(\left( T_1 \right)\) manera ineficiente (en comparación con la\(n \rightarrow \pi^*\) excitación de cetonas). Además, el\(S_1\) estado conduce a otras reacciones además de la isomerización que, en el caso del 1,2-difenileteno y otros hidrocarburos conjugados, producen productos cíclicos. Por ejemplo, cis-1,2-difenileteno irradiado en presencia de oxígeno da fenantreno por la secuencia de la Ecuación 28-8. La fotorreacción primaria es la ciclación a un intermedio de dihidrofenantreno\(6\), que, en presencia de oxígeno, se convierte en fenantreno:
La etapa de ciclación de la Ecuación 28-8 es una contraparte fotoquímica de las reacciones electrocíclicas discutidas en la Sección 21-10D. Se conocen muchas reacciones fotoquímicas similares de dienos y trienos conjugados, y son de gran interés porque, al igual que sus parientes térmicos, a menudo son estereoespecíficos pero tienden a exhibir estereoquímica opuesta a lo que se observa para reacciones térmicas formalmente similares. Por ejemplo,
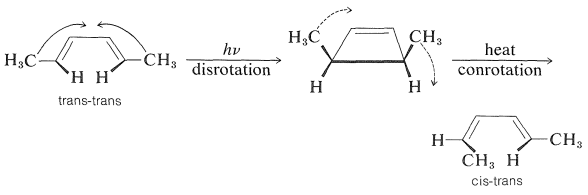
Estas reacciones son procesos concertados por\(4n\) electrones controlados por la simetría de los orbitales reaccionantes. La reacción térmica es más favorable con un estado de transición de Mobius (logrado por conrotación), mientras que la reacción fotoquímica es más favorable con un estado de transición de Huckel (logrado por disrotación).
Los dienos conjugados también experimentan reacciones fotoquímicas de cicloadición. Las cicloadiciones térmicas relacionadas de alcadienos se han discutido en las Secciones 13-3A, 21-10A y 21-10D, pero las reacciones térmicas y fotoquímicas frecuentemente dan diferentes productos cíclicos. El butadieno proporciona un excelente ejemplo de las diferencias:
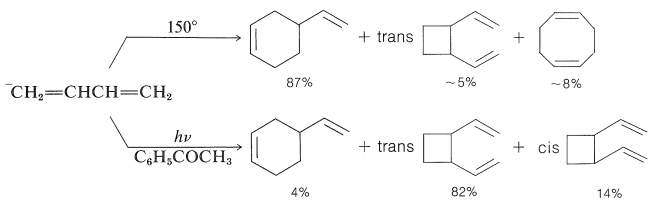
En la reacción térmica el aducto [4 + 2] o Diels-Alder es el producto principal, mientras que en la reacción fotoquímica dominan las cicloadiciones [2 + 2]. Debido a que las adiciones fotoquímicas son sensibilizadas por una cetona\(\ce{C_6H_5COCH_3}\), estas cicloadiciones ocurren a través del estado triplete de 1,3-butadieno y, como resultado, no es sorprendente que estas cicloadiciones sean paso a paso, no estereoespecíficas e involucren intermedios dirradicales.
Excitación:
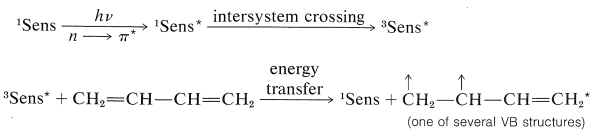
Cicloadición:
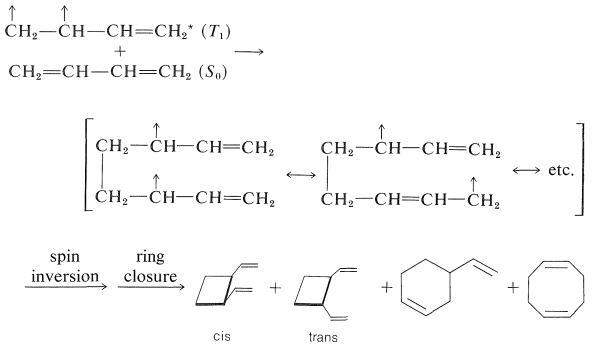
La irradiación directa de 1,3-butadieno con\(254\)\(\text{nm}\) luz produce ciclobuteno y pequeñas cantidades de biciclo [1.1.0] butano junto con dímeros.
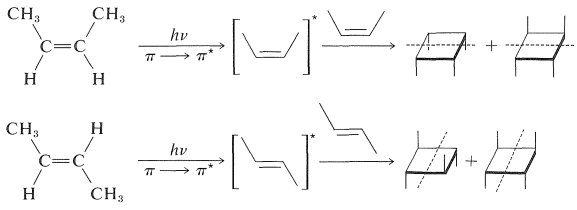
A diferencia de los dienos conjugados, los alquenos simples como el 2-buteno no reaccionan fácilmente por cicloadición fotosensibilizada. Pero formarán [2 + 2] cicloaductos en irradiación directa. Estas adiciones se producen por medio de un estado excitado singlete y son estereoespecíficas:
Una reacción relacionada, que no tiene precedentes en química térmica, es la cicloadición de un alqueno y un aldehído o cetona para formar un oxaciclobutano:
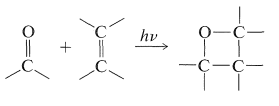
En este tipo de adición, el alqueno en estado fundamental\(\left( S_0 \right)\) reacciona con un estado excitado (usualmente\(T_1\)) del compuesto carbonilo por medio de un intermedio dirradical:
Reacciones de oxígeno singlete
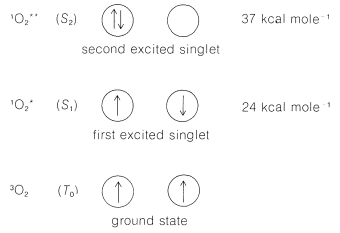
El estado fundamental del oxígeno molecular es inusual porque es un estado triplete. Dos electrones de espín paralelo ocupan\(\pi\) orbitales separados de igual energía (degenerados), como se muestra esquemáticamente en la Figura 28-5,\(^3\) Los siguientes dos estados electrónicos superiores ambos son estados singlete y se encuentran respectivamente\(24\) y\(37 \: \text{kcal mol}^{-1}\) por encima del estado fundamental. De esto podemos entender por qué el oxígeno ordinario tiene las propiedades de un dirradical y reacciona rápidamente con muchos radicales, como en la oxidación de la cadena radical de hidrocarburos (autooxidación; Secciones 15-10 y 16-9E):
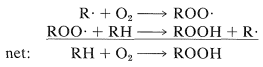
El oxígeno también apaga eficientemente los estados tripletes excitados de otras moléculas\(\left( ^3A^* \right)\) y, al aceptar energía triplete, es promovido a un estado singlete excitado. Observe que la orientación total del giro se conserva:
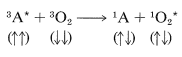
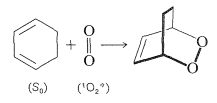
El oxígeno singlete es altamente reactivo hacia muchas moléculas orgánicas y formará productos oxigenados de adición o sustitución. Como ejemplo, los dienos conjugados reaccionan con oxígeno singlete para dar peróxidos mediante [4 + 2] cicloadición. Debido a que solo están involucrados estados singlete, esta adición es bastante análoga a las reacciones térmicas de Diels-Alder (Sección 21-10A):
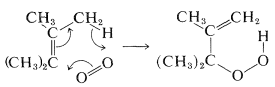
Si el alqueno o alcadieno tiene al menos un hidrógeno en el carbono adyacente al doble enlace, la reacción con oxígeno singlete puede dar hidroperóxidos. El mecanismo de esta reacción está relacionado con [4 + 2] cicloadiciones y se presume que ocurre a través de un estado de transición pericíclico de Huckel (ver Sección 21-10D):
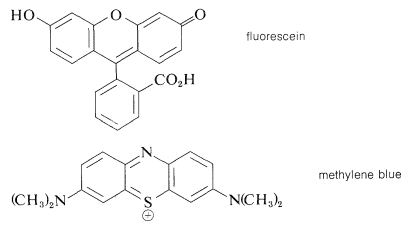
Muchas reacciones de este tipo se pueden lograr permitiendo que el hidrocarburo reaccione con el oxígeno en presencia de un tinte sensibilizante que absorbe fuertemente la luz visible. Los colorantes más utilizados para este propósito incluyen fluoresceína (y sus análogos clorados, eosina y rosa de bengala), azul de metileno y pigmentos de porfirina (como la clorofila).
El proceso global de oxigenación fotosensibilizada de un sustrato\(\left( \ce{A} \right)\) procede de las siguientes etapas:
\[^1\text{Sens} \overset{h \nu}{\longrightarrow} \: ^1\text{Sens}^* \longrightarrow ^3\text{Sens}^*\]
\[^3\text{Sens}^* + ^3\ce{O_2} \longrightarrow ^1\text{Sens} + ^1\ce{O_2}*\]
\[^1\ce{O_2}* + \ce{A} \longrightarrow \ce{AO_2}\]
El oxígeno singlete se puede producir químicamente así como por sensibilización fotoquímica. Existen varios métodos químicos disponibles, siendo uno de los más conocidos la reacción del hipoclorito de sodio con peróxido:
\[\ce{NaOCl} + \ce{H_2O_2} \rightarrow \ce{NaCl} + \ce{H_2O} + ^1\ce{O_2} \tag{28-9}\]
Un método alternativo de formación, que puede ser utilizado en disolventes orgánicos a bajas temperaturas, implica la descomposición térmica del ozónido de fosfito de trietilo (Ecuación 28-10):
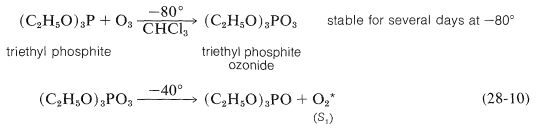
Independientemente de si el oxígeno singlete se forma química o fotoquímicamente, da productos similares en reacciones con alquenos.
Fotobiología
Las reacciones fotosensibilizadas de oxígeno son en gran medida perjudiciales para los organismos vivos. De hecho, el oxígeno singlete reacciona destructivamente con aminoácidos, proteínas y ácidos nucleicos. ¿Cómo se protege un organismo contra los efectos dañinos del oxígeno? No hay respuestas simples, pero las plantas verdes proporcionan una pista. La clorofila es un excelente colorante sensibilizante para el oxígeno singlete; sin embargo, las plantas verdes evidentemente no se ven perjudicadas por ello. Una razón puede ser que el oxígeno singlete es apagado muy eficientemente por otros pigmentos vegetales, especialmente los pigmentos carotenoides como el\(\beta\) -caroteno (Sección 2-1). Que este es el caso está indicado por el hecho de que las plantas mutantes incapaces de sintetizar caroteno son asesinadas rápidamente por el oxígeno y la luz.
Es bien conocido que la irradiación directa con luz ultravioleta es perjudicial para los organismos unicelulares. También se sabe que los ácidos nucleicos, ADN y ARN, son las dianas importantes del daño fotoquímico, y este conocimiento ha estimulado mucha investigación en el campo de la fotobiología con la esperanza de desentrañar la química involucrada.
Un resultado interesante y significativo es el hallazgo de que las bases de pirimidina de los ácidos nucleicos (uracilo, timina y citosina) son fotorreactivas y experimentan [2 + 2] cicloadiciones en irradiación con luz ultravioleta. La timina, por ejemplo, da un dímero de estructura\(7\):
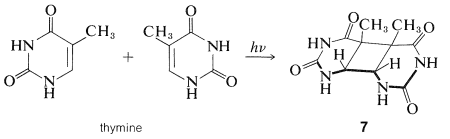
Experimentos comparables con los ácidos nucleicos han confirmado que la cicloadición de sus bases de pirimidina también ocurre con luz ultravioleta y entrecruza eficazmente las cadenas, proceso obviamente bastante contrario al funcionamiento del ADN (Sección 25-13B). Un aspecto notable y poco entendido de la fotobiología es el mecanismo de reparación y defensa que tanto las plantas como los animales poseen para minimizar los efectos dañinos de la radiación.
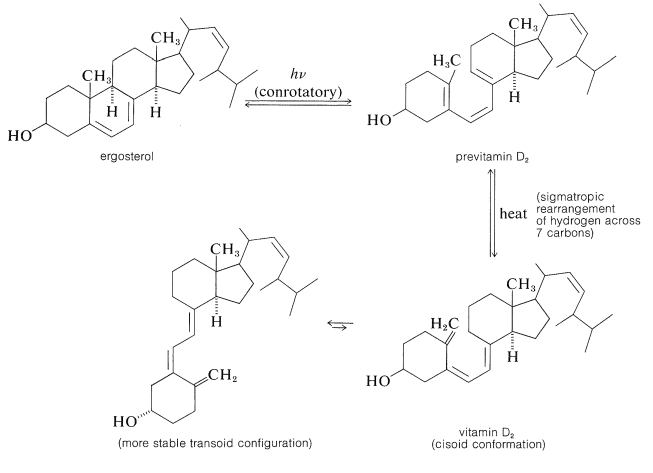
En el lado positivo, existen reacciones fotoquímicas que son esenciales para la salud humana. Uno de ellos es la formación de vitamina D (la vitamina antiracítica) por irradiación de ergosterol. Esta reacción fotoquímica es una abertura de anillo electrocíclico del anillo ciclohexadieno de ergosterol del tipo descrito en la Sección 28-2D. El producto, previtamina D 2, posteriormente se reorganiza térmicamente a vitamina D 2:
\(^1\)La unidad de einstein se define en la Sección 9-4.
\(^2\)Recibidor con G. Porter del Premio Nobel de Química en 1967 por trabajo sobre reacciones fotoquímicas.
\(^3\)Para un relato más detallado de la configuración electrónica del oxígeno molecular, véase M. Orkin y H. H. Jaffe, The importance of Antibonding Orbitals, Houghton Mifflin Co., Boston, 1967; o H. B. Gray, Chemical Bonds, W. A. Benjamin, Inc., Menlo Park, Calif., 1973.
Colaboradores y Atribuciones
- John D. Robert and Marjorie C. Caserio (1977) Basic Principles of Organic Chemistry, second edition. W. A. Benjamin, Inc. , Menlo Park, CA. ISBN 0-8053-8329-8. This content is copyrighted under the following conditions, "You are granted permission for individual, educational, research and non-commercial reproduction, distribution, display and performance of this work in any format."