
1.22: Proteínas
- Page ID
- 74280

Secuenciación de un péptido
La última vez analizamos las características estructurales de los aminoácidos y el enlace peptídico que une los aminoácidos individuales para formar proteínas y péptidos. También aprendimos sobre la secuencia (orden) en la que las unidades de aminoácidos se unen en péptidos. Hoy estudiaremos las formas en que se puede descubrir la secuencia específica de un péptido y los métodos que se utilizan para sintetizar dicho péptido.
Lo primero que se hace para determinar la secuencia de un péptido es averiguar qué aminoácidos están presentes y en qué proporciones. Esto es muy parecido a comenzar el proceso de determinar la estructura de un compuesto orgánico determinando las proporciones de átomos como carbono, hidrógeno y oxígeno. Esto se hace hidrolizando los enlaces peptídicos que mantienen el péptido unido usando HCl como catalizador ácido. (El mecanismo es muy parecido a la hidrólisis de éster catalizada por ácido).
Este “análisis de aminoácidos” nos dice cuáles son los bloques de construcción en el péptido, pero no nos dice nada sobre su secuencia, el orden en que se unen. Esta información se pierde cuando se hidrolizan los enlaces peptídicos que conservan esa secuencia. Incluso con tan pocos como dos aminoácidos, hay dos secuencias posibles. Consideremos un dipéptido que el análisis de aminoácidos nos da gly y ala. Cualquiera de estos podría ser el extremo N, por lo que el dipéptido podría ser gly-ala o ala-gly. El problema 18.6 en Brown te da algo de experiencia con un pentapéptido, y las cosas se vuelven más complejas rápidamente a medida que aumenta el número de unidades de aminoácidos en el péptido.
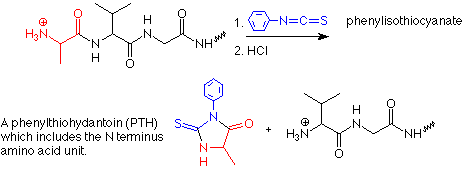
La siguiente etapa es determinar qué aminoácidos ocupan las posiciones N terminal y C terminal en el péptido. La determinación del extremo N se realiza comúnmente mediante un proceso llamado degradación de Edman. La química se describe de la siguiente manera:
Esta reacción se puede entender si buscamos algunas analogías que nos ayuden a aplicar los patrones que usamos en el pasado. El grupo -N=C=S se asemeja a una molécula de CO 2 (O=C=O) en que el átomo de carbono está conectado a dos átomos electronegativos por un doble enlace (sigma y pi). Sabemos por la reacción de los reactivos de Grignard con CO 2 que el nucleófilo ataca al carbono en CO 2, por lo que podemos esperar el mismo tipo de patrón en la degradación de Edman. El nucleófilo es el grupo NH 2 libre en el extremo N del péptido, formado por la pérdida de un protón desde el NH 3 + hasta alguna base no especificada. Como hemos visto con otras reacciones de los grupos NH 2, a este paso le sigue un desplazamiento de protones.
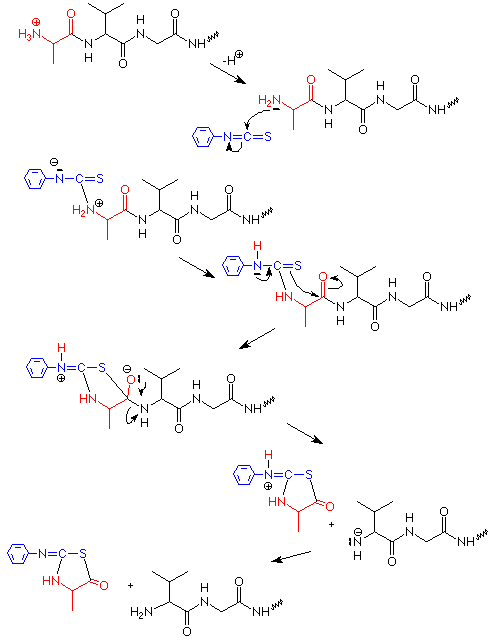
El producto de la adición de N y H al doble enlace C=N tiene un átomo de azufre nucleófilo localizado justo al alcance del carbono carbonilo en el otro extremo del aminoácido N-terminal. El ataque de este azufre a ese grupo carbonilo es seguido por la salida del grupo NH del siguiente aminoácido. Esto escinde el enlace peptídico entre el aminoácido N terminal y el siguiente aminoácido. La reorganización adicional de protones produce un isómero de la feniltiohidantoína. Este isómero se convierte en la feniltiohidantoína durante el tratamiento con HCl y se identifica la feniltiohidantoína. Dado que la feniltiohidantoína incluye el grupo R del aminoácido N terminal, la identificación de la feniltiohidantoína también identifica el aminoácido N-terminal.
El otro producto de la degradación de Edman también es un péptido, es el péptido original menos el aminoácido N terminal original. Ahora tiene un nuevo aminoácido N terminal, que era adyacente al aminoácido N terminal en el péptido original. El nuevo péptido también puede ser sometido a degradación de Edman. Cuando se hace esto, aprendemos la identidad del segundo aminoácido (desde el extremo N terminal) del péptido original y obtenemos nuevamente un péptido que ahora es dos aminoácidos más corto que el original. En principio, la repetición de esta secuencia nos permitiría ejecutar sucesivas degradaciones de Edman, recortando un aminoácido N terminal con cada degradación, y así aprender la secuencia completa de un péptido o proteína. En la práctica, tal proceso es práctico solo para aproximadamente 20 a 40 aminoácidos.
Dado que los pasos de laboratorio en una degradación de Edman son muy repetitivos —lo mismo se hace en cada ciclo, ha sido posible automatizar este proceso. Los “secuenciadores de proteínas” controlados por computadora son comunes en los laboratorios de bioquímica.
Secuencias superpuestas
Cuando se van a secuenciar péptidos y proteínas mayores de 20 a 40 unidades de aminoácidos, primero se rompen en fragmentos más pequeños, ya sea por hidrólisis parcial química o enzimática. Aquí hay un ejemplo de cómo se podría usar la hidrólisis química parcial.

Por supuesto, la angiotensina II (un péptido implicado en la regulación de la presión arterial) es lo suficientemente pequeña como para que un secuenciador de proteínas pueda dar su secuencia directamente, pero el ejemplo ilustra la forma en que los fragmentos pueden superponerse para dar una secuencia completa para una proteína más grande. La secuencia completa de una proteína se llama su estructura primaria
Síntesis de Péptidos
Cuando se ha obtenido una secuencia para un péptido, se puede prestar atención a su síntesis. Hay dos problemas que resolver en la síntesis de un péptido. Una es desarrollar un método para hacer el enlace peptídico que no dañe nada más en el péptido. Esto se llama “acoplamiento” de los dos aminoácidos. La otra es asegurarse de que los aminoácidos se agregan al péptido en la secuencia apropiada.
La clave del primer problema es convertir el grupo carboxilato de O - un aminoácido en un mejor grupo de salida. Hemos visto algo similar cuando hemos convertido un OH en un Cl como hicimos los cloruros de acilo reactivos. Esto se hace mediante el uso de un reactivo llamado diciclohexil carbodiimida, o DCC para abreviar. DCC funciona uniéndose a la O - y convirtiéndola en un buen grupo de salida - bueno porque tiene muchos átomos electronegativos que pueden ayudar a estabilizar la carga negativa a medida que sale. Se dice que un aminoácido que tiene un buen grupo lábil está “activado”.
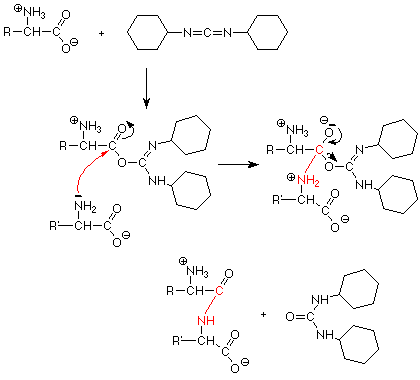
La reacción de acoplamiento real ocurre cuando el grupo amino del aminoácido “a la derecha” en la secuencia ataca el carbono carbonilo del aminoácido “activado” y el DCC sale con el oxígeno al que está unido. Como es habitual, hay algunos turnos de protones necesarios para ordenar las cosas.
La segunda cuestión, la adición de aminoácidos en la secuencia deseada, puede ilustrarse considerando la síntesis de un dipéptido como Ala-Gly. Si simplemente mezclamos cantidades iguales de glicina y alanina y realizamos una reacción de acoplamiento DDC, obtendremos glicinas que reaccionan con glicinas para dar Gly-Gly, alaninas reaccionando con alaninas para dar Ala-Ala, y glicinas reaccionando con alaninas de dos maneras para dar Ala-Gly y Gly-Ala. Esto es un desastre y sería mejor desarrollar un enfoque más específico.
Para ello necesitamos disponer las cosas de manera que solo uno de los dos grupos carboxilo y solo uno de los dos grupos amino sean libres para participar en la reacción de acoplamiento. Esto se hace mediante el uso de grupos protectores. Si hacemos un grupo amino en una amida, es mucho menos reactivo como nucleófilo. Esto se puede hacer por reacción con un cloruro de ácido.
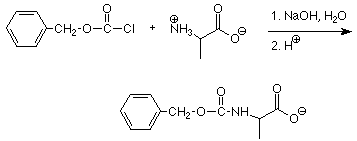
Los grupos carboxilo normalmente están protegidos por conversión a un éster bencílico. Esta reacción es una esterificación de Fischer
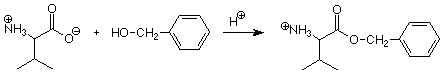
Después del acoplamiento, los grupos protectores se pueden eliminar por hidrogenación.
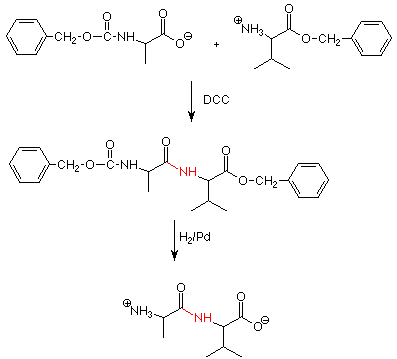
Esta es una reacción específica en la que solo se rompen los enlaces entre los grupos bencilo y los oxígenos, por lo que el enlace amida que acabamos de hacer por acoplamiento no se ve afectado. (El grupo carbonilo en el grupo amino anteriormente protegido se pierde como CO 2). Se han desarrollado esquemas de protección más sofisticados en los que la protección N o la protección O pueden eliminarse selectivamente.
En la práctica, las proteínas se sintetizan ahora mediante técnicas de biología molecular en las que se aísla el gen que codifica la secuencia de aminoácidos y se utiliza para dirigir la síntesis de la proteína por una bacteria o una levadura.
Estructura secundaria, terciaria y cuaternaria
Cuando estamos pensando en la secuencia de un péptido es conveniente pensarlo como una cadena que está estirada, los péptidos son más comúnmente enrollados (hélice alfa, Brown p 518) o plegados (lámina beta, Brown p 519). Estas formas se llaman estructura secundaria de un péptido o proteína y se mantienen en su lugar principalmente por enlaces de hidrógeno. Recordemos que los enlaces de hidrógeno son mucho más débiles que los enlaces covalentes, pero lo suficientemente fuertes como para resistir la ruptura por temperaturas suaves. Los enlaces de hidrógeno son interacciones atractivas entre el extremo positivo de dipolos como los enlaces N-H y O-H y ubicaciones cargadas negativamente como los pares de electrones no compartidos en átomos como oxígeno o nitrógeno. En los péptidos son comúnmente los enlaces N-H de una amina y los oxígenos de los grupos carbonilo los que participan en los enlaces de hidrógeno.
Las regiones de hélice alfa o lámina beta a menudo se combinan mediante patrones de plegamiento adicionales que conforman la estructura terciaria de una proteína. Las proteínas estructurales como la queratina o la fibroína a menudo tienen grandes regiones de hélice alfa y forman fibras. Las enzimas son globulares con mayor frecuencia con aminoácidos hidrófilos en el exterior y aminoácidos hidrófobos plegados hacia el medio.
Las proteínas individuales a menudo se combinan en racimos, que pueden incluir moléculas no proteicas como el hemo (Fig 18.14, p 522 en Brown). A menudo tales combinaciones son necesarias para que la proteína pueda llevar a cabo su función biológica. Dichos conglomerados constituyen la estructura cuaternaria de la proteína.
Los experimentos han establecido que la estructura primaria de una proteína es suficiente, por sí misma, para determinar cómo se plegará y combinará con otras proteínas para hacer las estructuras secundarias, terciarias y cuaternarias apropiadas. No está claro cómo sucede esto, y esta es un área de estudio activo.