
1.25: Eliminación - E2 y E1
- Page ID
- 74231

S N 1 Energía y Cinética
La última vez vimos una visión general de los mecanismos de sustitución nucleofílica de haluros de alquilo. Examinamos uno de estos, el mecanismo S N 2 en detalle. Hoy examinaremos el otro, el mecanismo S N 1, y luego veremos las reacciones de eliminación, la mayor competencia por las sustituciones. Aquí está el esquema del mecanismo S N 1:
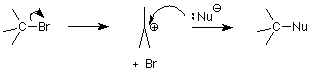
Recordando lo que significaba el “2" en S N 2 —que la reacción era de segundo orden, dos moléculas tuvieron que colisionar para proporcionar la energía de activación necesaria para alcanzar y pasar por el estado de transición— podemos adivinar que el “1" en S N 1 significa que solo una molécula necesita ser “activada” en para llegar al estado de transición. Esa molécula es el haluro de alquilo. El paso crítico en este mecanismo es el primer paso, en el que se rompe el enlace entre el átomo de carbono y el grupo saliente halógeno. El estado de transición para esta etapa tiene el enlace estirado lo suficiente como para que el ion haluro esté equilibrado entre salir como un ion cloruro o bromuro estable o deslizarse de nuevo en un enlace covalente.
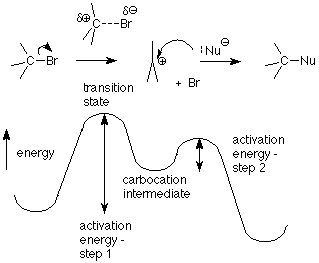
(La energía requerida para romper este enlace proviene de colisiones aleatorias con el solvente sin que el solvente reaccione). La energía de activación para el primer paso es mayor que para el segundo paso, por lo que la velocidad de la reacción está controlada por la cantidad de moléculas que pasan por el primer paso. Una vez que una molécula pasa por el primer paso, puede reaccionar rápidamente en el segundo paso. Llamamos al paso que es más lento el “paso determinante de la tasa”. Observe que el paso determinante de la velocidad para esta reacción no involucra al nucleófilo. Eso significa que cambiar la concentración del nucleófilo no afecta la velocidad. La única concentración que afecta la velocidad es la concentración del haluro de alquilo. Así tenemos una reacción de primer orden:
Tasa = k [RX]
Esto también significa que la fuerza del nucleófilo —su capacidad para usar su par de electrones para hacer un enlace— no es importante para determinar qué tan rápido va la reacción. No está involucrado en el paso determinante de la tasa, por lo que no tiene ningún efecto sobre la energía de ese estado de transición.
Efecto de la Estructura del Grupo Alquílico
¿Qué afecta la energía del estado de transición determinante de la tasa? Si examinamos su estructura con más detalle, notamos que hay una carga positiva considerable en el átomo de carbono y el enlace carbono-halógeno está casi roto.
El enlace casi roto nos dice que el efecto de cambiar el halógeno es el mismo que lo fue para la reacción de S N 2:
RI > RBr > RCl > RF
El grado sustancial de carga positiva sobre el carbono es importante para explicar cómo la estructura del grupo alquilo afecta la velocidad. Un gran número de experimentos han establecido que el orden de reactividad para los haluros de alquilo en el mecanismo S N 1 es:
terciario > secundario > primario > metilo
Esto es justo lo contrario del orden para la reacción de S N 2. El resultado de este contraste es que los haluros de alquilo terciario utilizan consistentemente la vía S N 1. Los haluros primarios y de metil alquilo utilizan la vía S N 2. Para los haluros de alquilo secundarios es posible cualquier vía, y tenemos que mirar otra información para tomar una decisión.
El orden de reactividad nos indica que el estado de transición para la reacción de SN 1 de un haluro de alquilo terciario tiene una energía menor que el estado de transición para un haluro de alquilo secundario correspinding. Quizás la forma más fácil de entender esto es reconocer que el estado de transición, con su carga positiva sustancial sobre el carbono, es muy parecido al intermedio carbocatiónico que se forma en el primer paso. Los cambios que hacen que el carbocatión intermedio, con su carbono cargado positivamente, sea más estable también harán que el estado de transición, con su carbono cargado casi positivamente, sea más estable.
Lo principal que determina la energía del carbocatión intermedio es que solo tiene seis electrones en su caparazón de valencia. Es un electrófilo, un ácido de Lewis y una molécula seriamente deficiente en electrones. Cuanta más densidad de electrones se pueda desplazar hacia el carbono cargado positivamente, menor será la energía. En un carbocatión terciario, hay tres átomos de carbono, cada uno unido a otros tres átomos, conectados al carbono del carbocatión deficiente en electrones. Esto suma 18 electrones en los enlaces adyacentes al carbocatión, todos los cuales pueden desplazarse ligeramente para ayudar a neutralizar la carga positiva. Contraste esto con la situación en una meti carbocatión. Aquí no hay electrones de valencia que no sean los que sostienen los hidrógenos al carbono, por lo que hay un suministro muy pobre de electrones para ayudar a disminuir la energía del carbocatión.
Si bien esta discusión se ha centrado en el carbocatión intermedio con su carga positiva completa, los mismos principios se aplican en el caso de la carga positiva parcial en el estado de transición. Cuantos más electrones haya en el vecindario cercano, más estable será el estado de transición. Hay más electrones disponibles en los átomos de carbono unidos a un centro terciario de carbocatión que en los hidrógenos unidos a un centro de carbocatión de metilo.
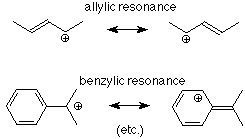
Si hay enlaces pi involucrados con un carbono unido al carbono carbocatión, la energía se reduce aún más. Recordemos que los electrones pi están menos unidos que los electrones sigma, por lo que son más fáciles de avanzar hacia la deficiencia de electrones. Esto se puede simbolizar en términos de resonancia:
El efecto de esto es que los haluros de alquilo que tienen enlaces carbono-carbono pi ubicados a un átomo del carbono que lleva el halógeno son bastante reactivos en las reacciones de S N 1.
Resultado estereoquímico
El producto de una reacción de S N 1 se forma en la segunda etapa. Este paso comienza con el carbocatión intermedio, así que veamos su estructura para ver qué podemos aprender de él. Primero, observe la distinción entre un estado intermedio y un estado de transición. Recuerde que un estado de transición está en una energía máxima. Cualquier ligero cambio hará que caiga de su precaria perca y se convierta en el producto o el reactivo de su paso. En contraste, un intermedio está en un mínimo de energía por lo que la mayoría de los cambios pequeños solo lo empujan un poco hacia arriba una colina desde su situación de menor energía. Se necesita una colisión sustancial para proporcionarle la energía necesaria para alcanzar un estado de transición para que pase y se convierta en un producto. Si la energía de activación es bastante pequeña, como lo es con el segundo paso del mecanismo S N 1, la reacción bien puede ser rápida, pero no ocurre con cada pequeño meneo como lo hace con un estado de transición.
Otra forma de decir esto es que un estado de transición tiene una vida útil muy, muy corta, mientras que un intermedio puede existir por micro o milisegundos, lo que es mucho tiempo y brinda oportunidades para muchas colisiones con otras moléculas.
Un resultado de esto es que el carbocatión intermedio “vive” el tiempo suficiente para que un nucleófilo se acerque a él en cualquiera de las caras de la molécula. El carbocatión es plano con ángulos de 120 o entre sus tres enlaces. Eso significa que es trigonal como el carbono de un grupo carbonilo y es sp 2 hibridado. El orbital “vacío” que podemos asociar con las características electrofílicas del carbocatión es un orbital p.
Si el carbocatión intermedio tiene la oportunidad de participar en muchas colisiones con nucleófilos potenciales, existe la misma probabilidad de que un nucleófilo ataque en un lóbulo u otro de la órbita p. Si hay tres grupos diferentes unidos al centro de carbocatión, el nucleófilo proporcionará un cuarto y generará un carbono estereogénico. Se esperarán cantidades iguales de los dos enantiómeros para que la mezcla de productos no sea ópticamente activa.
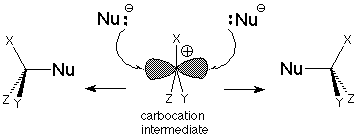
Se trata de una situación algo idealizada, ya que en la práctica el grupo saliente de haluro se encuentra “merodeando” cerca de un lóbulo de la órbita p. Esto facilita la reacción con el nucleófilo en el otro lóbulo, por lo que suele haber alguna inversión neta. El resultado estereoquímico del S N 1 no es tan claro como el de la inversión del mecanismo S N 2, pero sigue siendo una característica distintiva del mecanismo, uno que puede ser utilizado para decidir qué mecanismo está operando en una reacción particular.
Solvolisis
El hecho de que el nucleófilo no esté involucrado en la etapa de determinación de la velocidad de una reacción de S N 1 también significa que procede bien con un nucleófilos relativamente débiles. Cuando el nucleófilo es también el solvente, la reacción se llama “solvolisis”. Los disolventes como el agua y los alcoholes son particularmente útiles aquí, ya que proporcionan tanto un par nucleofílico de electrones en el átomo de oxígeno del grupo OH como un disolvente bastante polar que ayuda a estabilizar el estado de transición fuertemente polar. Este último efecto es muy parecido a la forma en que un disolvente polar disuelve una sustancia muy polar como la sal rodeando sus iones cargados con moléculas de disolvente polar.
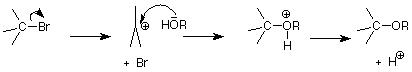
Este proceso se asemeja a la forma en que un alcohol o agua podría atacar el carbono electrófilo de un grupo carbonilo después de que un ácido hubiera agregado un H + al oxígeno. Recuerda que en estas reacciones el átomo nucleófilo neutro ataca primero; luego se pierde el H +.
Un resumen de las diferencias importantes entre los mecanismos S N 1 y S N 2 se encuentra en el Cuadro 7.5 (Brown, p 189).
Competencia con Eliminación
Una de las cosas que tratamos la última vez fue el hecho de que los nucleófilos también son bases de Lewis. Un resultado de esto es que el mismo átomo o grupo puede atacar a un carbono en una reacción de S N 1 o S N 2 —comportándose como nucleófilo— o atacar a un átomo de hidrógeno, comportándose como una base de Lewis. Este último ataque puede llevar a una reacción de eliminación. Analizaremos las reacciones de eliminación con más detalle en una semana más o menos, pero podemos examinarlas útilmente como competidores para las reacciones de S N 1 y S N 2 en este momento.
Cuando aprendimos por primera vez sobre la síntesis de éter de Williamson, aprendimos que funciona mejor cuando el haluro de alquilo es primario. Ahora entendemos que como característica del mecanismo S N 2, los haluros de alquilo primarios reaccionan más rápido que los haluros de alquilo secundario o terciario. Sin embargo, una persona paciente podría sugerir que incluso una reacción lenta de S N 2 podría tener éxito si estuviéramos dispuestos a esperar un rato. Lo que derrota a esta estrategia es que el ion alcóxido también puede reaccionar como base. Esta reacción de eliminación (una reacción E 2 para referencia futura) es lo suficientemente rápida como para utilizar el haluro de alquilo secundario o terciario mucho antes de que la reacción de S N 2 mucho más lenta produzca cualquier cantidad útil de producto.
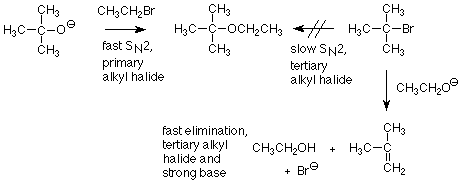
Las reacciones de eliminación son siempre competidores potenciales para las reacciones de sustitución. Los factores clave que nos alertan de situaciones favorables a las eliminaciones son:
- Un haluro de alquilo que es lento en reacciones de S N 2, es decir, haluros de alquilo terciario y secundario.
- La presencia de una base fuerte como un ion alcóxido o hidróxido.
Estas condiciones típicamente producirán mucho más producto de eliminación (un alqueno) que producto de sustitución.
¿Podemos usar una vía S N 1 para evitar esta dificultad? Sí, y la clave aquí es que dado que la velocidad de una reacción de S N 1 no es sensible a la concentración o fuerza del nucleófilo podemos evitar las bases fuertes que promueven la eliminación. La solvolisis de un haluro de alquilo terciario usando un alcohol como nucleófilo y disolvente puede hacer que un éter sea muy efectivo.
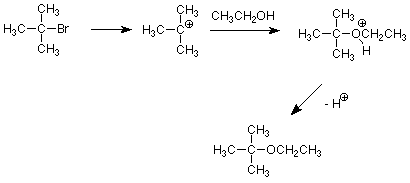
Por supuesto, esto solo es posible cuando el nucleófilo deseado se puede usar como disolvente como es el caso de los alcoholes y el agua.
S N 1 y S N 2 Reacciones de Alcoholes
Para terminar hoy, revisemos algunas reacciones de los alcoholes y veamos si podemos usar el patrón S N 1 o S N 2 para entenderlas un poco mejor. Recordemos que un método útil para convertir un alcohol en un haluro de alquilo fue tratar el alcohol con el haluro de hidrógeno, particularmente cuando el alcohol era terciario:
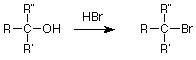
Esto parece una sustitución nucleofílica y dado que el grupo alquilo es terciario, la vía S N 1 a través de un intermedio de carbocatión parece una buena suposición. También podemos identificar al nucleófilo como el ion bromuro (Br -), pero ¿qué pasa con el grupo saliente? La respuesta obvia es que el OH -sirve como grupo saliente, pero esto es preocupante ya que estaríamos haciendo una base fuerte en presencia de un ácido. La solución aparece cuando recordamos que los pares de electrones no compartidos en el oxígeno del alcohol también son bases débiles. Pueden aceptar un protón (H +) del ácido fuerte HBr. Cuando esto se hace, el grupo de salida es agua, una base débil.
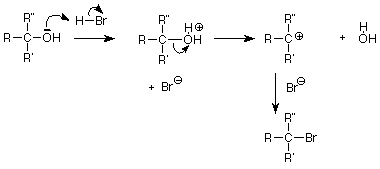
Cuando el grupo OH de un alcohol es reemplazado por otro nucleófilo, podemos estar seguros de que el grupo OH primero se convierte en un buen grupo saliente antes de que se rompa el enlace C-O. Esa es la función del H + en un ácido, la parte SO 2 del cloruro de tionilo y el fósforo en PBr 3. No nos preocuparemos por los detalles de estos procesos, pero podemos notar que la necesidad de convertir al grupo OH en un buen grupo de salida es la misma si la reacción es S N 1 como esperaríamos para un alcohol terciario o S N 2 como esperaríamos para un alcohol primario.