
1.4: Análisis de reactividad polar
- Page ID
- 70212

El par de electrones p en un enlace C=C puede estar distribuido de manera desigual si el enlace p lleva o está conjugado con un grupo funcional. Es razonable aproximar la distribución del par de electrones en, por ejemplo, el enol o enolato π-enlace como teniendo abundancia de electrones en el β-carbono ya que un sustituyente hidroxilo activa el β-carbono hacia la formación de enlaces con un electrófilo de carbono. Así, los grupos funcionales estabilizan los centros vecinos de abundancia de electrones (centros nucleofílicos) y los centros de deficiencia de electrones (centros electrófilos). Por ejemplo, el carbono carbonilo de una cetona es electrófilo mientras que el carbono α- a un carbonilo es potencialmente nucleófilo. 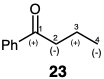 La nucleofilia real en este carbono se obtiene cuando este carbono se conjuga con el carbono carbonilo como el enol o enolato correspondiente. De manera similar, el enlace enona C-C π-es electrófilo en el carbono β, mientras que el γ-carbono es potencialmente nucleofílico, estando disponible la nucleofilia real por conversión al enol o enolato correspondiente. Así, la activación polar proporcionada por un grupo funcional puede extenderse a centros de carbono remotos por conjugación. Esta posibilidad está indicada para 23. En la discusión subsiguiente, los centros de reactividad electrofílica o nucleofílica real o potencial serán designados como (+) y (-) respectivamente en contraposición a los centros de carga positiva o negativa, los cuales serán designados como\(\oplus\) y\(\ominus\).
La nucleofilia real en este carbono se obtiene cuando este carbono se conjuga con el carbono carbonilo como el enol o enolato correspondiente. De manera similar, el enlace enona C-C π-es electrófilo en el carbono β, mientras que el γ-carbono es potencialmente nucleofílico, estando disponible la nucleofilia real por conversión al enol o enolato correspondiente. Así, la activación polar proporcionada por un grupo funcional puede extenderse a centros de carbono remotos por conjugación. Esta posibilidad está indicada para 23. En la discusión subsiguiente, los centros de reactividad electrofílica o nucleofílica real o potencial serán designados como (+) y (-) respectivamente en contraposición a los centros de carga positiva o negativa, los cuales serán designados como\(\oplus\) y\(\ominus\).
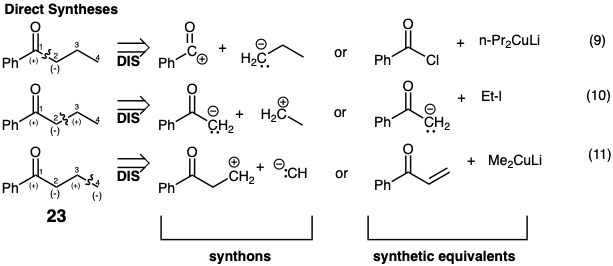
Consideremos todas las estrategias sintéticas posibles para la síntesis directa del grupo butirilo de butirofenona (23) a partir de dos fragmentos mediante una reacción polar que explota la activación polar proporcionada por el grupo carbonilo o cualquier precursor de grupo funcional que confiera electrofilicidad al mismo átomo de carbono. Hay tres posibles desconexiones C-C del objetivo 23. Las tres posibles estrategias de conexión C-C se resumen en las ecuaciones 9-11. Las estrategias pueden considerarse primero en términos generales al representar los precursores polares reactivos requeridos como sintones. El grupo carbonilo en 23 proporciona reactividad electrofílica en el carbono 1 permitiendo una síntesis por creación polar del enlace 1-2 por reacción con un nucleófilo de tres carbonos. El grupo carbonilo en 23 también proporciona potencialmente reactividad nucleofílica en el carbono 2 permitiendo una síntesis por creación polar del enlace 2-3 por reacción con un electrófilo de dos carbonos. El grupo carbonilo en 23 también proporciona potencialmente reactividad electrofílica en el carbono 3 permitiendo la síntesis por creación polar del enlace 3-4 por reacción con un nucleófilo de un carbono. Por lo general, se reconocen los sintones requeridos pero solo se consideran explícitamente los equivalentes sintéticos apropiados de estos sintones. Para cada una de las síntesis anteriores son posibles una gran variedad de equivalentes sintéticos alternativos. Solo es necesario que el precursor electrófilo elegido tenga un nivel de funcionalidad una unidad mayor que el carbono correspondiente en la diana (o tenga un enlace C=C conjugado con un grupo funcional activante electrofílicamente) y que el precursor nucleófilo tenga un nivel de funcionalidad una unidad menor que el carbono correspondiente en el objetivo para lograr una síntesis directa de la diana.
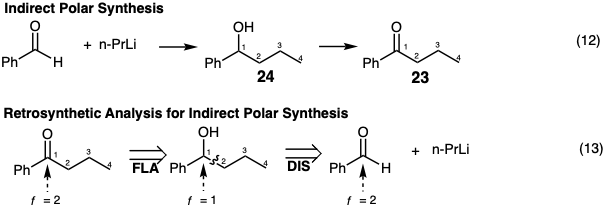
Una síntesis indirecta del objetivo también puede ser razonable. Por ejemplo, 23 podría prepararse mediante la adición de N-prli a benzaldehído produciendo un intermedio 24 con el esqueleto de carbono de la diana 23. El ajuste posterior del nivel de funcionalidad por oxidación luego entrega la cetona 23 (ecuación 12). Retrosintéticamente tal estrategia requiere el reconocimiento de la posibilidad de que el benzaldehído sea un precursor electrófilo fácilmente disponible de la porción benzoílo de 23. Sin embargo, dado que el nivel de funcionalidad de este electrófilo es el mismo en el carbono incipiente 1 que en el objetivo, este último no puede producirse directamente a partir del benzaldehído en un proceso de formación de enlaces C-C polares. Más bien, la primera dislocación del objetivo debe ser el ajuste de su nivel de funcionalidad (FLA) previo a la desconexión polar en la segunda dislocación (ecuación 13).
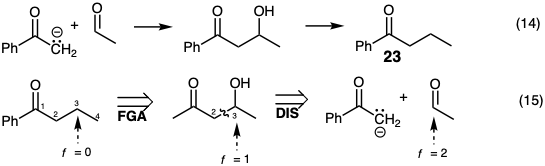
Topologicamente, la estrategia de la ecuacion 13 esta relacionada con la de la ecuacion 9. En la ecuación 14 se esboza una síntesis indirecta de 23 por una estrategia relacionada topológicamente con la de la ecuación 10. Retrosintéticamente tal estrategia requiere el reconocimiento de la posibilidad de que el acetaldehído sea un electrófilo de dos carbonos fácilmente disponible. Sin embargo, dado que el nivel de funcionalidad de este electrófilo es dos unidades mayor que el carbono incipiente 3 en 23, una reacción de formación de enlaces polares con un nucleófilo de carbono conduce a un producto en el que el nivel de funcionalidad en este carbono es una unidad superior al requerido para 23. Por lo tanto, la primera dislocación del objetivo debe ser el ajuste de su nivel de funcionalidad (aquí adición de grupo funcional, FGA, un caso especial de FLA) previo a la desconexión polar en la segunda dislocación (ecuación 15).
Reacciones polares regioselectivas
Es importante reconocer que todas las estrategias consideradas anteriormente son hipotéticas. La reacción sintética deseada entre los intermedios elegidos puede no ser la única vía de reacción disponible. Por ejemplo, los sintones deslocalizados son inherentemente ambientes; poseen varios centros de reactividad. Así, el enolato en la ecuación 14 es un nucleófilo ambidente que puede reaccionar con un electrófilo ya sea en el oxígeno carbonilo o α-carbono. De manera similar, el electrófilo conjugado 25 puede reaccionar con un nucleófilo ya sea en el carbono carbonilo (adición 1,2-) o en el carbono β (adición 1,4- o Michael). Así, 25 es un electrófilo ambiente. Para ser sintéticamente útil, la formación de enlaces debe realizarse en la posición deseada de un nucleófilo o electrófilo ambiente, la reacción debe ser regioselectiva.

Elementos de Control de Reactividad
Ahora hemos visto que las estrategias indirectas, que implican dislocaciones de una diana que no reducen directamente la complejidad molecular, pueden ser deseables porque: (1) permiten una dislocación posterior de la diana que simplifica de manera eficiente la complejidad molecular, (2) explican un inicio fácilmente disponible materiales que tienen un alto nivel de complejidad molecular, o (3) explican ciertos electrófilos o nucleófilos fácilmente disponibles que tienen niveles de funcionalidad que son inapropiados para la síntesis conectiva directa C-C de una diana. Las estrategias indirectas pueden ser deseables por otras razones. Así, algunos átomos o grupos de átomos pueden explotarse para controlar reacciones sintéticas alterando la selectividad. Nos referiremos a dichos fragmentos moleculares como elementos de control.
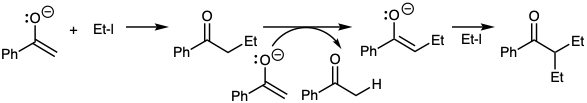
Por ejemplo, la alquilación de cetonas como en la ecuación 10 a menudo da como resultado la polialquilación debido a transferencias rápidas de protones de la cetona del producto al enolato de partida. La fuerte basicidad de los enolatos de cetona también puede resultar en la abstracción de protones de haluros de alquilo (β-eliminación) en lugar de la sustitución nucleofílica. La adición de un grupo carboetoxi proporciona un nucleófilo menos básico menos reactivo que puede alquilarse con buen rendimiento (ecuación 16). Tal grupo carboetoxilo a menudo se conoce como un grupo activador ya que activa la molécula hacia la abstracción de protones. Sin embargo, quizás sea más significativo que este grupo desactive al nucleófilo resultante convirtiéndolo en un reactivo más selectivo. Después de que haya cumplido su propósito, el elemento de control debe ser removido. Retrosintéticamente, la conveniencia de explotar un elemento de control requiere la adición de ese elemento (CEA) en la primera dislocación del objetivo previo a la reacción que se pretende controlar que entonces es la segunda dislocación del objetivo (ecuación 17).
Objetivos difuncionales
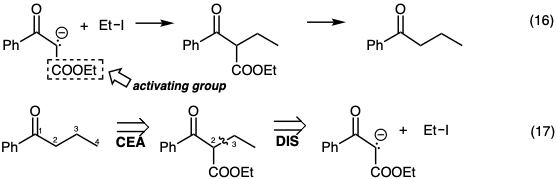
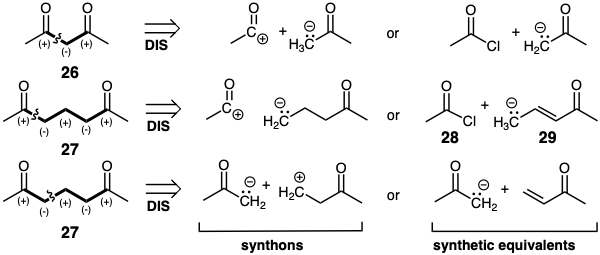
Las síntesis polares de difuncionales difuncionales mediante estrategias que explotan la activación polar proporcionada por ambos grupos funcionales para lograr la formación de enlaces C-C pueden dividirse en dos categorías: aquellas cuyo esqueleto de carbono puede ensamblarse directamente con la funcionalidad requerida y aquellas que no pueden. Las síntesis conectivas C-C que generan directamente dianas difuncionales con la funcionalidad requerida son posibles si ambos grupos funcionales imparten la misma reactividad polar real o potencial a los átomos que conectan los grupos funcionales. Nos referiremos a tales grupos funcionales y a los átomos que los conectan como circuitos consonantes. Por ejemplo, 26 y 27 contienen circuitos consonantes. La síntesis conectiva directa C-C de dianas difuncionales constantes se puede lograr mediante: (a) adición conjugada a un electrófilo cuyo nivel de instauración es una unidad mayor que el de la diana, o (b) sustitución nucleófila o adición a un electrófilo cuyo nivel de funcionalidad es una unidad mayor que el del objetivo. Cabe señalar que, si bien el esqueleto de carbono se ensambla directamente con el nivel de funcionalidad correcto en las estrategias anteriores, no siempre es posible lograr una síntesis directa de dianas difuncionales consonantes con el nivel de instauración requerido. Así, la reacción de 28 con 29 generará un producto con el esqueleto de carbono de 27 pero con mayor instauración. Así, la dislocación 27 ⇒ 28 + 29 debe ser un proceso de dos pasos.
También son posibles estrategias indirectas para ensamblar objetivos difuncionales consonantes. Por lo tanto, durante la formación del enlace C-C, se puede generar un intermedio que no tenga el nivel de funcionalidad adecuado para la diana deseada. Sin embargo, si bien los niveles de funcionalidad de los precursores pueden ser diferentes a los del objetivo, la funcionalidad en los equivalentes sintéticos de esos precursores es electrónicamente la misma que la funcionalidad de la diana difuncional consonante. Nos referiremos a la funcionalidad en precursores cuya nucleofilia o electrofilicidad es la misma que la del objetivo —pero cuyo nivel puede diferir— como funcionalidad relacionada con la diana.
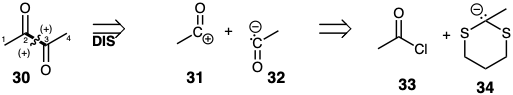
La disonancia de reactividad polar está presente en dianas difuncionales si la reactividad polar impartida a los átomos de conexión por un grupo funcional es revertida por el otro. Nos referiremos a tales grupos funcionales y a los átomos que los conectan como circuitos disonantes. Por ejemplo, 30 contiene un circuito disonante. La síntesis de dianas difuncionales disonantes nunca se puede lograr mediante rutas conectivas C-C que explotan directamente la activación polar proporcionada por ambos grupos funcionales. Por ejemplo, la desconexión polar de 30 en el enlace 2,3-debe generar un sintón acil-nucleófilo 32. Pero el grupo carbonilo generalmente proporciona reactividad electrofílica como en 31 y no reactividad nucleofílica en el carbono carbonilo. Se conocen equivalentes sintéticos tales como 34 de dichos sintones anormales, es decir, equivalentes de carbanión acilo. Contienen una funcionalidad que está relacionada con la del objetivo pero en la que se enmascara la reactividad polar habitual de la funcionalidad diana y se estabiliza la reactividad polar opuesta.
La reactividad polar normal de los grupos funcionales puede enmascararse por conversión a derivados no reactivos. 11 Se dice que el grupo funcional está enmascarado, bloqueado o protegido en tales derivados. Los grupos funcionales no reactivos así creados se denominan grupos enmascarantes o protectores. Dichos grupos son ejemplos de elementos de control de reactividad. Existe una subclase de grupos enmascarantes que no solo bloquean la reactividad polar normal de un grupo funcional sino que también facilitan la reactividad polar opuesta. Tal inversión de la reactividad polar de un grupo funcional se ha denominado umpölung. 12 Por ejemplo, la reactividad electrofílica normal del carbono carbonilo en acetaldehído puede transformarse en reactividad nucleofílica por desprotonación del tioacetal derivado 35. El grupo ditioacetal no solo enmascara la electrofilicidad del precursor carbonilo, sino que también facilita la desprotonación del carbono carbonilo estabilizando el carbanión derivado. Así, el anión 34 es un equivalente sintético del sintón carbanión acilo 32. La acilación de 34 suministraría 36 a partir de la cual la diana disonante 30 puede derivarse por hidrólisis. Obsérvese que el grupo carbonilo en el material de partida acetaldehído se explota indirectamente, es decir, después de la inversión de su reactividad polar habitual, para la generación polar de un enlace C-C diana.
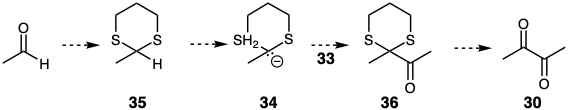
La desconexión polar de 37 en el enlace 2,3-debe generar un sintón 32 o 38 con reactividad polar invertida mientras que la desconexión en el enlace 3-4 debe generar un sintón 44 con reactividad polar invertida. Cabe señalar que los equivalentes sintéticos de sintones con reactividad polar invertida, por ejemplo 34, 39, 41 y 45, por definición, son moléculas disonantes. Así, por ejemplo, mientras que el carbono carbonílico enmascarado tiene reactividad nucleofílica en 41, la nucleofugacidad de los grupos tiofenilo también hace que este carbono sea potencialmente electrofílico. En este caso las reactividades polares opuestas son conferidas por un único grupo funcional, el ditioacetal, un grupo funcional que puede proporcionar activación tanto nucleofílica como electrófila al mismo carbono.
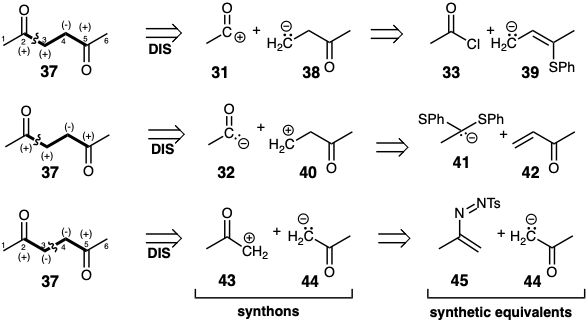
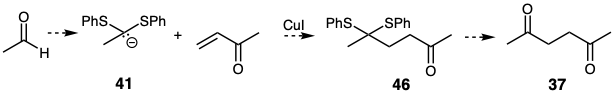
Un equivalente de acil carbanión 41 está disponible por desprotonación del derivado di (feniltio) acetal del acetaldehído. La adición conjugada del carbanión 41 a la metil vinil cetona podría producir el tiocetal 46 del cual se obtendría la diana disonante 37 por hidrólisis.
Una gran variedad de equivalentes sintéticos de “sintones umpoled” están disponibles. 13 Aunque incorporan funcionalidad enmascarada con reactividad invertida, no se preparan necesariamente por umpolung del grupo funcional relacionado con la diana. Por ejemplo, el equivalente sintético 48 del sintón 47 es un equivalente de ion acetaldehído enolonio. Reacciona con el enolato de ciclohexanona para suministrar 49 del cual se obtiene el objetivo disonante 50 tras la hidrólisis. 14
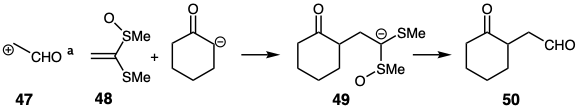
El equivalente de ion acetaldehído enolonio 48 se puede obtener a partir de cloruro de metil magnesio y disulfuro de carbono por desprotonación del ditioacetato intermedio seguido de S-metilación y luego por oxidación selectiva con ácido m-cloroperbenzoico (MCPBA). 14
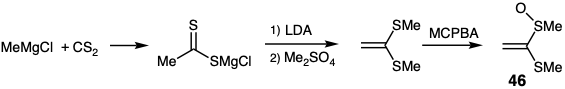
Dianas disonantes de precursores disonantes
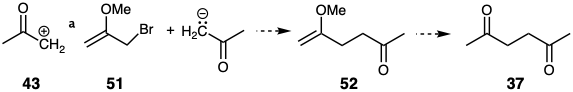
La síntesis polar conectiva C-C de dianas difuncionales disonantes también se puede lograr mediante secuencias de múltiples etapas empleando reacciones polares que explotan la reactividad polar proporcionada por solo uno de los dos grupos funcionales en un precursor disonante. Por ejemplo, la alilación del enolato de acetona con bromuro de 2-metoxialilo (51), un equivalente sintético del sintón de acetona enolonio (43), seguido de la hidrólisis del éter de enol intermedio 52 podría proporcionar la diana difuncional disonante 37. Aunque 51 se prepara a partir de acetona, la reactividad polar proporcionada por el grupo carbonilo del precursor de acetona no está involucrada en la reacción de 51 con nucleófilos. Más bien, el grupo carbonilo —como derivado no reactivo— es un transeúnte inocente. La reactividad polar requerida para la formación de enlaces C-C es proporcionada por un segundo grupo funcional diana no relacionado, es decir, el grupo bromo. También, debe reconocerse que 51 es en sí mismo una molécula difuncional disonante. La reacción del enolato de acetona con 51 proporciona otro ejemplo de un principio general: las dianas disonantes están disponibles por reacciones conectivas C-C polares de un precursor disonante. Así, 51 es una molécula difuncional disonante.
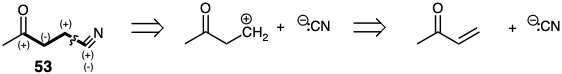
Como se señaló anteriormente para los ditioacetales, algunos grupos funcionales no solo proporcionan reactividad electrófila en el carbono al que están unidos sino que también facilitan la generación de carbaniones (es decir, reducción) en ese carbono. Dado que esto permite reactividad tanto electrófila como nucleofílica en el carbono funcional o cualquier carbono conjugado con él, nos referiremos a él como un grupo funcional bifílico. Por ejemplo, el =N confiere electrofilicidad al carbono en un nitrilo y también facilita la desprotonación de HC=N para conferir nucleofilia al mismo carbono. Así, aunque el carbono nitrilo en 53 es electrófilo y 53 es por lo tanto un objetivo difuncional disonante, el ion cianuro es un equivalente nucleófilo estable del sintón de carbanión nitrilo. La diana disonante 53 está disponible directamente mediante la adición del conjugado polar de cianuro a metil vinil cetona.
En resumen, las dianas disonantes pueden construirse mediante secuencias multietapa empleando reacciones polares que explotan: (a) inversión de la reactividad polar de un grupo funcional en la diana; (b) solo uno de los dos grupos funcionales en un precursor disonante para proporcionar reactividad polar; (c) un bifílico grupo funcional.
Sintesis no polares de dianas disonantes
Las dianas difuncionales disonantes a menudo se preparan mediante reacciones no polares conectivas C-C directas, tales como oxidaciones, reducciones, procesos de desplazamiento de enlaces pericíclicos o adiciones de radicales libres. Así, el acoplamiento reductivo implica la unión de dos centros electrofílicos acompañados de la adición de un par de electrones como en la reacción pinacol de acetona para producir el producto disonante difuncional pinacol.
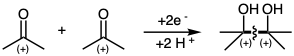
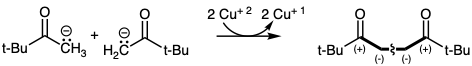
El acoplamiento oxidativo implica la unión de dos centros nucleofílicos acompañados por la eliminación de un par de electrones. La dicetona disonante 54 se obtiene tras el acoplamiento oxidativo del enolato de pinacolona.
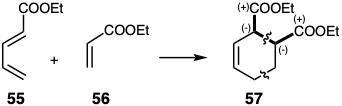
La generación de dianas disonantes mediante procesos de desplazamiento de enlaces pericíclicos es posible ya que la orientación de tales reacciones está controlada por solapamiento orbital p que no necesariamente se corresponde con la reactividad polar. Por ejemplo, el diéster disonante 57 es el producto principal de la cicloadición de 55 con 56.
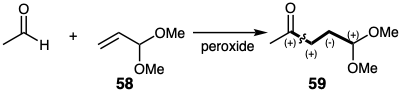
La reacción en cadena de radicales libres entre acetaldehído y acetal 58 para generar 59 ejemplifica otra ruta conectiva C-C no polar para productos difuncionales disonantes.
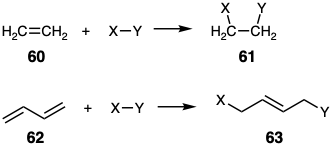
Las dianas difuncionales disonantes también están disponibles mediante procesos conectivos no C-C tales como la adición de átomos electronegativos X e Y a ambos carbonos de un enlace C=C. Nos referiremos a tales reacciones como adiciones dioxidativas ya que ambos carbonos están oxidados. Las adiciones dioxidativas también pueden ocurrir con los polienos. Tales reacciones, que llamaremos adiciones 1, n-dioxidativas, siempre generan difuncionalidad disonante. Así, la conversión de 60 en 61 implica la adición 1,2-dioxidativa mientras que la conversión de 62 a 63 es una adición 1,4-dioxidativa.
Desconexión de Bonos C=C
Las dislocaciones retrosintéticas de una diana sintética que implican la desconexión de un doble enlace carbono-carbono, es decir, desconexiones dobles, generalmente corresponden a secuencias sintéticas de múltiples etapas. No hay reacciones polares que generen dos enlaces entre dos átomos de carbono en un solo paso. (Obsérvese que las dimerizaciones de carbenos son cicloadiciones, que por definición, generan dos enlaces en un solo paso.) Por lo tanto, si se va a explotar la activación polar, se debe hacer una doble conexión durante la síntesis en dos etapas: la primera, una unión polar; la segunda, una eliminación. La etapa de eliminación generalmente implica la pérdida de HX, XY o MX donde X e Y son grupos que son más electronegativos que el carbono, mientras que M es cualquier grupo que es más electropositivo que el carbono. Así, retrosintéticamente, la desconexión a través de un enlace C=C requiere una adición como la primera dislocación de la diana.
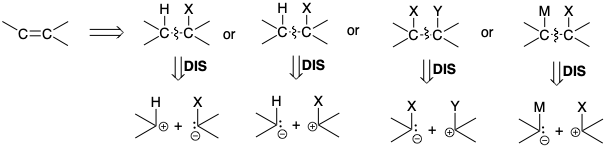
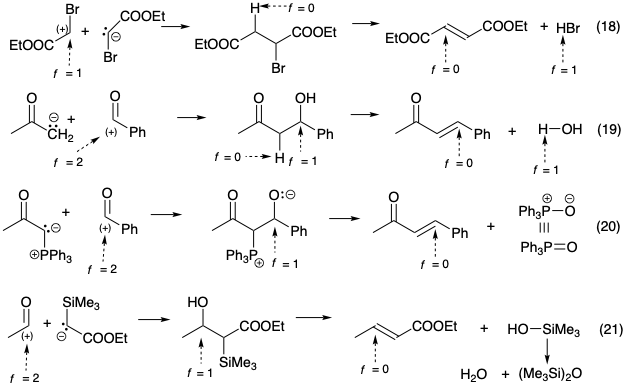
La síntesis esbozada en la ecuación 18 es un ejemplo representativo del primer enfoque. Aquí un grupo activador electrófilo de nivel de funcionalidad = 1 reside en cada carbono. Las síntesis esbozadas en las ecuaciones 19-21 son ejemplos representativos de cada una de las tres últimas estrategias. En cada caso, el nivel de funcionalidad del sintón electrófilo disminuye en dos al ir a la diana C=C. El nivel de funcionalidad del sintón nucleofílico va de -1, -2 o 0 para las ecuaciones 19, 20 y 21 respectivamente en los precursores a 0 en los productos. También tenga en cuenta que los segundos pasos en las ecuaciones 18 y 19 requieren la oxidación de un hidrógeno (desprotonación), y que el segundo paso en la ecuación 20 es una eliminación direductora.
Fallas Estratégicas
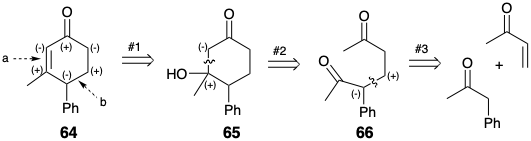
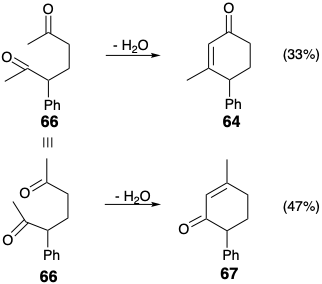
La activación polar proporcionada por un solo grupo funcional en una diana puede explotarse numerosas veces durante una síntesis para facilitar varias etapas de conexión C-C. Por ejemplo, el grupo carbonilo de la 2-ciclohexen-1-ona 64 podría usarse para proporcionar la reactividad nucleófila y electrófila requerida para generar dos conexiones C-C en esta diana (enlaces a y b) correspondientes a las dislocaciones #2 y #3 (etapas sintéticas 1 y 2) en una estrategia para síntesis de 64 a partir de metil vinil cetona y fenil acetona. La primera dislocación de la diana, la adición de agua al enlace C=C muestra la necesaria intervención de un intermedio durante la doble conexión (etapas sintéticas 2 y 3) correspondientes a la conversión 65 a 64. Este plan sintético también proporciona un ejemplo de un diseño sintético significativamente defectuoso ya que la δ-dicetona 66 intermedia se cicla de dos maneras diferentes, solo una de las cuales proporciona el producto deseado. La 2-ciclohexen-1-ona isomérica 67 puede ser incluso el producto principal de esta reacción. 15
Teoría y Práctica
Además de las fallas en lograr la regioselectividad requerida durante las reacciones de adición de nucleófilos o electrófilos multidentados, o las reacciones de moléculas con varios grupos funcionales similares, la eliminación planificada de la funcionalidad activadora, o la instauración, así como la introducción, eliminación o la interconversión de la funcionalidad por oxidación, reducción, metátesis, etc., puede no ser factible debido a limitaciones de reacciones conocidas y/o limitaciones impuestas por la reactividad característica de una diana sintética particular. El análisis de reactividad polar sirve meramente para generar sistemáticamente un conjunto de estrategias sintéticas. Estos deben evaluarse posteriormente en términos de la disponibilidad de reacciones selectivas adecuadas y materiales de partida apropiados.