
11.2: Espectroscopia infrarroja (IR)
- Page ID
- 76719
Introducción
Las energías fotónicas asociadas con el infrarrojo (de 1 a 15 kcal/mol) no son lo suficientemente grandes como para excitar electrones, pero pueden inducir la excitación vibracional de átomos y grupos unidos covalentemente.
Los enlaces covalentes en las moléculas no son barras o varillas rígidas, como los que se encuentran en los kits de modelos moleculares, sino que son más como resortes rígidos que pueden estirarse y doblarse. La naturaleza móvil de las moléculas orgánicas se observó en el capítulo relativo a los isómeros conformacionales. Ahora debemos reconocer que, además de la fácil rotación de grupos alrededor de enlaces simples, las moléculas experimentan una amplia variedad de movimientos vibracionales, característicos de sus átomos componentes. En consecuencia, prácticamente todos los compuestos orgánicos absorberán la radiación infrarroja que corresponde en energía a estas vibraciones. Los espectrómetros infrarrojos, similares en principio al espectrómetro UV-Visible descrito en otra parte, permiten a los químicos obtener espectros de absorción de compuestos que son un reflejo único de su estructura molecular.
Espectroscopía Vibracional
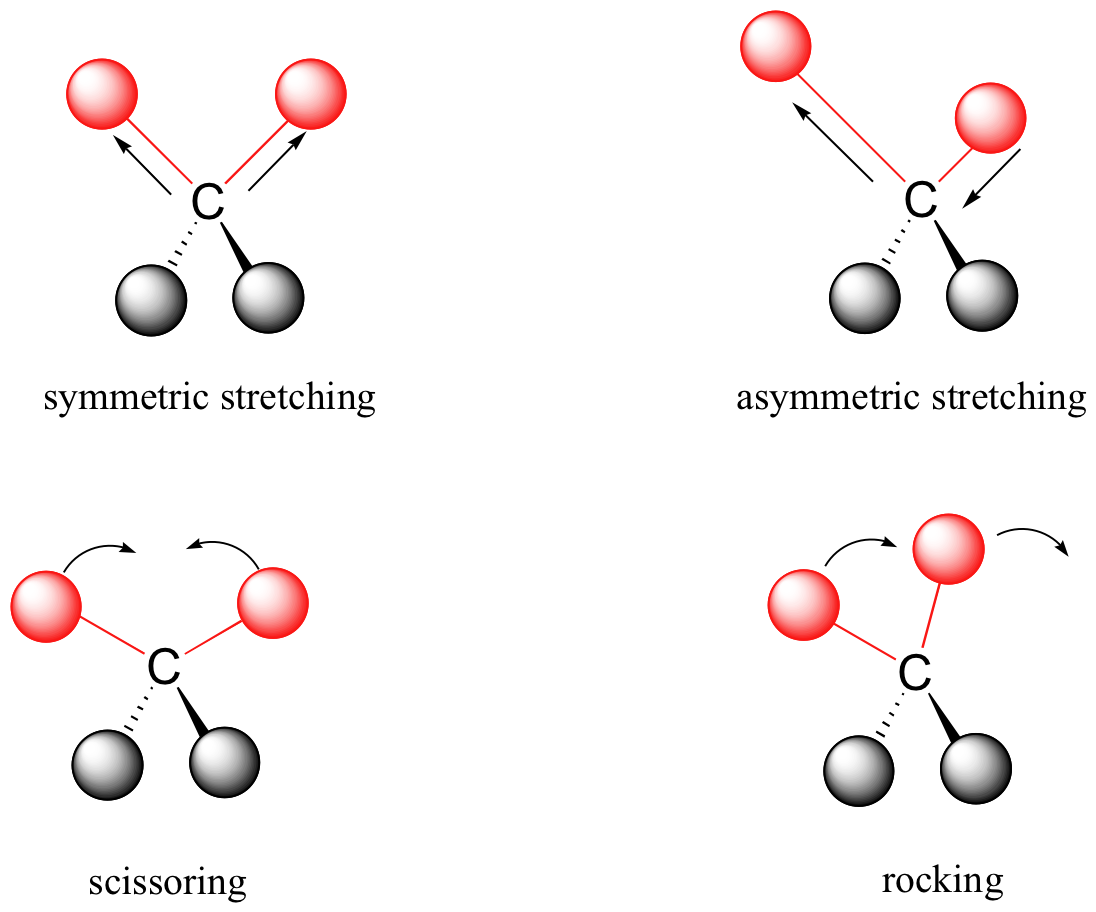
Una molécula compuesta por n-átomos tiene 3n grados de libertad, seis de los cuales son traslaciones y rotaciones de la propia molécula. Esto deja 3n-6 grados de libertad vibratoria (3n-5 si la molécula es lineal). A los modos vibracionales se les suele dar nombres descriptivos, como estiramiento, flexión, tijera, balanceo y torsión. La molécula de formaldehído de cuatro átomos, cuyo espectro en fase gaseosa se muestra a continuación, proporciona un ejemplo de estos términos. Si un modelo de formaldehído con bola y varilla no se muestra a la derecha del espectro, presione el botón view ball&stick model a la derecha. Se esperan seis vibraciones fundamentales (12 menos 6), y estas han sido asignadas a las absorciones del espectro. Para ver la molécula de formaldehído mostrar una vibración, haga clic en uno de los botones debajo del espectro, o haga clic en uno de los picos de absorción en el espectro.
Los enlaces covalentes en las moléculas orgánicas no son barras rígidas, sino que se comportan más como resortes. A temperatura ambiente, las moléculas orgánicas están siempre en movimiento, ya que sus enlaces se estiran, se doblan y se retuercen. Estas vibraciones complejas se pueden desglosar matemáticamente en modos vibratorios individuales, algunos de los cuales se ilustran a continuación.
La energía de la vibración molecular se cuantifica en lugar de continua, lo que significa que una molécula solo puede estirarse y doblarse a ciertas frecuencias 'permitidas'. Si una molécula se expone a radiación electromagnética que coincide con la frecuencia de uno de sus modos vibracionales, en la mayoría de los casos absorberá energía de la radiación y saltará a un estado de energía vibracional más alto, lo que esto significa es que la amplitud de la vibración aumentará, pero la la frecuencia vibracional seguirá siendo la misma. La diferencia de energía entre los dos estados vibracionales es igual a la energía asociada a la longitud de onda de radiación que se absorbió. Resulta que es la región infrarroja del espectro electromagnético la que contiene frecuencias correspondientes a las frecuencias vibracionales de los enlaces orgánicos.
Un espectro IR
Utilizaremos una muestra de cetonas para ilustrar este proceso. La muestra se irradia con luz infrarroja y el enlace carbonilo absorberá específicamente luz con esta misma frecuencia, que por las ecuaciones 4.1 y 4.2 corresponde a una longitud de onda de 5.83 x 10 -6 m y una energía de 4.91 kcal/mol. Cuando el enlace carbonilo absorbe esta energía, salta hasta un estado vibratorio excitado.
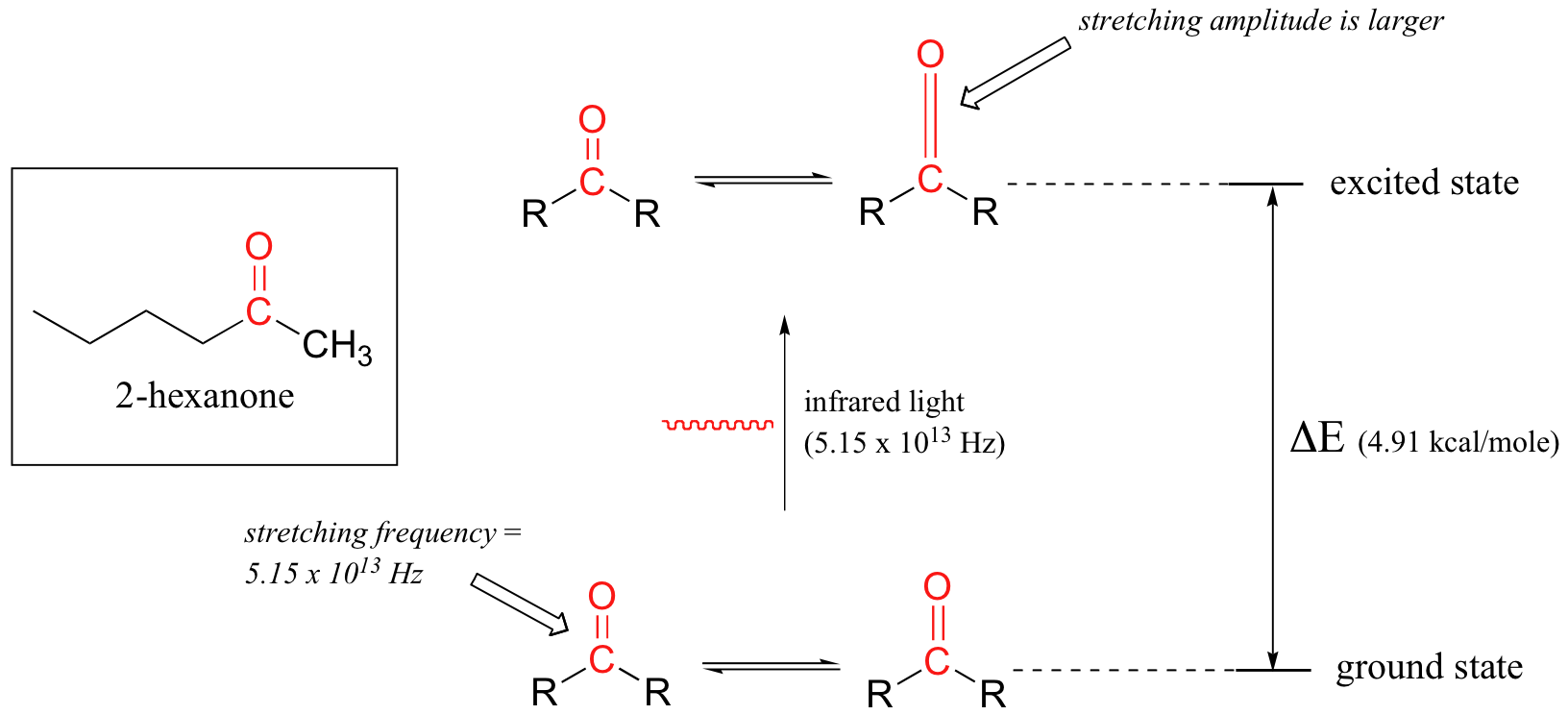
El valor de Δ E - la diferencia de energía entre los estados vibracionales de baja energía (tierra) y alta energía (excitada) - es igual a 4.91 kcal/mol, lo mismo que la energía asociada a la frecuencia de luz absorbida. La molécula no permanece en su estado vibratorio excitado por mucho tiempo, sino que libera rápidamente energía al ambiente circundante en forma de calor, y regresa al estado fundamental.
Con un instrumento llamado espectrofotómetro infrarrojo, podemos 'ver' esta transición vibracional. En el espectrofotómetro, la luz infrarroja con frecuencias que van desde aproximadamente 10 13 a 10 14 Hz se pasa a través de nuestra muestra de ciclohexano. La mayoría de las frecuencias pasan a través de la muestra y son registradas por un detector en el otro lado.
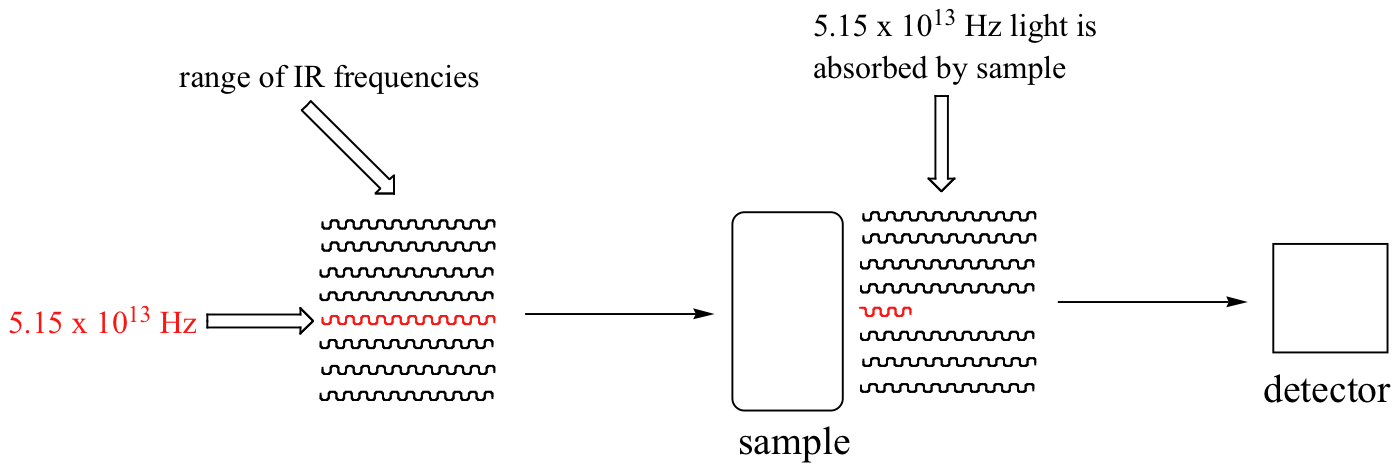
Nuestra frecuencia de estiramiento de carbonilo de 5.15 x 10 13 Hz, sin embargo, es absorbida por la muestra de 2-hexanona, por lo que el detector registra que la intensidad de esta frecuencia, después de haber pasado por la muestra, es algo menor que el 100% de su intensidad inicial.
Las vibraciones de una molécula de 2-hexanona no se limitan, por supuesto, al simple estiramiento del enlace carbonilo. Los diversos enlaces carbono-carbono también se estiran y se doblan, al igual que los enlaces carbono-hidrógeno, y todos estos modos vibracionales también absorben diferentes frecuencias de luz infrarroja.
El poder de la espectroscopia infrarroja surge de la observación de que diferentes grupos funcionales tienen diferentes frecuencias de absorción características. El enlace carbonilo en una cetona, como vimos con nuestro ejemplo de 2-hexanona, normalmente absorbe en el rango de 5.11 - 5.18 x 10 13 Hz, dependiendo de la molécula. El triple enlace carbono-carbono de un alquino, por otro lado, absorbe en el rango 6.30 - 6.80 x 10 13 Hz. Por lo tanto, la técnica es muy útil como medio para identificar qué grupos funcionales están presentes en una molécula de interés. Si pasamos luz infrarroja a través de una muestra desconocida y encontramos que absorbe en el rango de frecuencia carbonilo pero no en el rango de alquino, podemos inferir que la molécula contiene un grupo carbonilo pero no un alquino.
Ahora, veamos algunos resultados reales de experimentos de espectroscopia IR. A continuación se muestra el espectro IR para 2-hexanona.
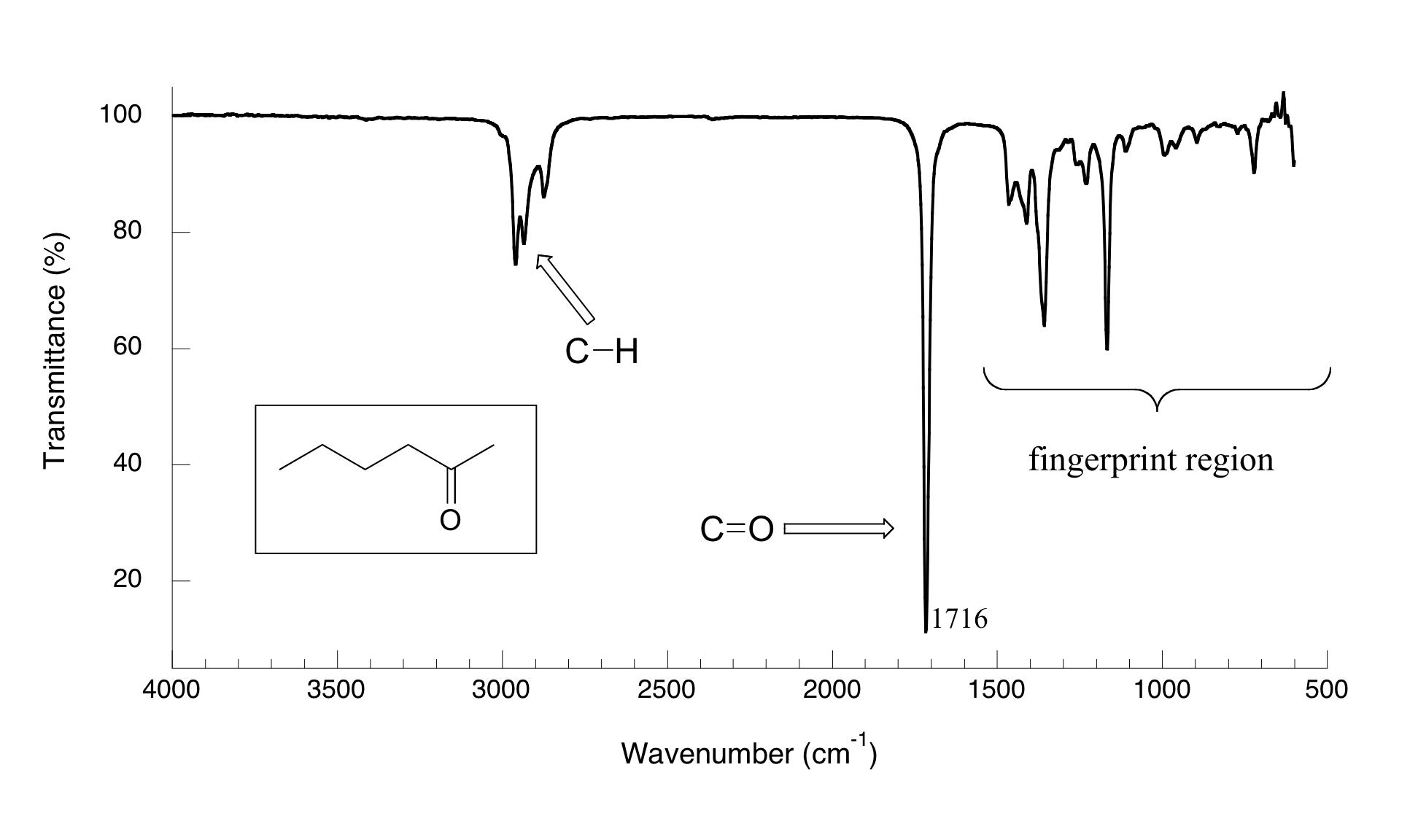
Hay una serie de cosas que hay que explicar para que entiendas qué es lo que estamos viendo. En el eje horizontal vemos longitudes de onda IR expresadas en términos de una unidad llamada número de onda (cm -1), que nos dice cuántas ondas caben en un centímetro. En el eje vertical vemos '% transmitancia', lo que nos dice con qué intensidad se absorbió la luz en cada frecuencia (100% de transmitancia significa que no se produjo absorción a esa frecuencia). La línea continua traza los valores de% de transmitancia para cada longitud de onda: los 'picos' (que en realidad apuntan hacia abajo) muestran regiones de fuerte absorción. Por alguna razón, es típico en la espectroscopia IR reportar valores de número de onda en lugar de longitud de onda (en metros) o frecuencia (en Hz). El eje vertical “al revés”, con picos de absorbancia apuntando hacia abajo en lugar de hacia arriba, es también una curiosa convención en espectroscopia IR. ¡No queremos facilitarte las cosas!
A la derecha se proporciona una calculadora para interconvertir estos valores de frecuencia y longitud de onda. Simplemente ingrese el valor a convertir en el cuadro correspondiente, presione "Calcular" y el número equivalente aparecerá en la casilla vacía.
Los espectros infrarrojos se pueden obtener de muestras en todas las fases (líquida, sólida y gaseosa). Los líquidos generalmente se examinan como una película delgada intercalada entre dos placas de sal pulidas (tenga en cuenta que el vidrio absorbe la radiación infrarroja, mientras que el NaCl es transparente). Si se usan solventes para disolver sólidos, se debe tener cuidado para evitar oscurecer regiones espectrales importantes por absorción de solventes. Se utilizan comúnmente disolventes perclorados como tetracloruro de carbono, cloroformo y tetracloroeteno. Alternativamente, los sólidos pueden incorporarse en un disco delgado de KBr, prepararse a alta presión o mezclarse con un poco de líquido no volátil y triturarse hasta obtener una pasta (o mull) que se unta entre placas de sal.
Frecuencia - Convertidor de longitud de onda |
|
|---|---|
| Frecuencia en cm -1 Longitud de onda en μ |
|
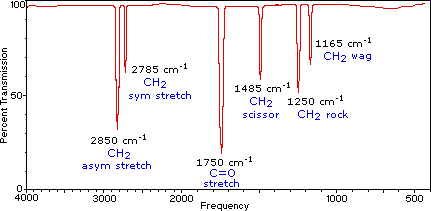
Espectro infrarrojo de formaldehído en fase gaseosa, H 2 C=O |
||
|---|---|---|
 |
||
|
Ball&Stick Modelo Spacefill Modelo Stick Modelo Motion Off |
|
La frecuencia exacta a la que ocurre una vibración dada está determinada por las fuerzas de los enlaces involucrados y la masa de los átomos componentes. Para una discusión más detallada de estos factores Haga clic aquí. En la práctica, los espectros infrarrojos normalmente no muestran señales de absorción separadas para cada uno de los modos vibracionales fundamentales 3n-6 de una molécula. El número de absorciones observadas puede ser incrementado por interacciones aditivas y sustractivas que conducen a la combinación de tonos y connotaciones de las vibraciones fundamentales, de la misma manera que interactúan las vibraciones sonoras de un instrumento musical. Además, el número de absorciones observadas puede disminuir por simetría molecular, limitaciones del espectrómetro y reglas de selección espectroscópica. Una regla de selección que influye en la intensidad de las absorciones infrarrojas, es que se debe producir un cambio en el momento dipolar para que una vibración absorba energía infrarroja. Las bandas de absorción asociadas con el estiramiento del enlace C=O suelen ser muy fuertes porque en ese modo se produce un gran cambio en el dipolo.
Algunas tendencias generales:
- Las frecuencias de estiramiento son más altas que las frecuencias de flexión correspondientes. (Es más fácil doblar un enlace que estirarlo o comprimirlo.)
- Los enlaces al hidrógeno tienen frecuencias de estiramiento más altas que las de los átomos más pesados.
- Los enlaces triples tienen frecuencias de estiramiento más altas que los dobles enlaces correspondientes, que a su vez tienen frecuencias más altas que los enlaces simples. (Excepto los enlaces a hidrógeno).
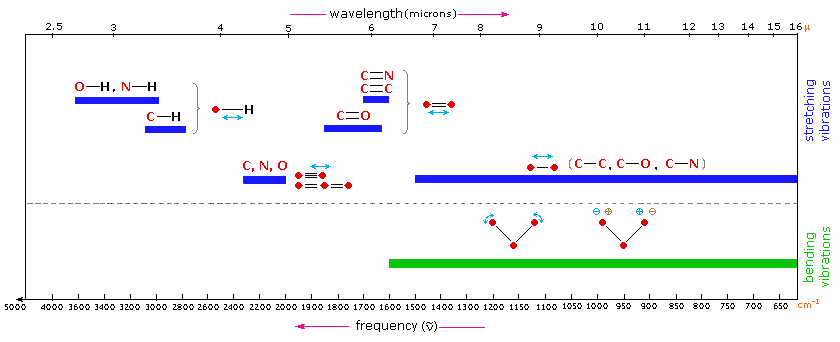
Las regiones generales del espectro infrarrojo en las que se observan diversos tipos de bandas vibratorias se describen en la siguiente tabla. Tenga en cuenta que las secciones de color azul por encima de la línea discontinua se refieren a vibraciones de estiramiento, y la banda de color verde debajo de la línea abarca vibraciones de flexión. La complejidad de los espectros infrarrojos en la región de 1450 a 600 cm -1 dificulta la asignación de todas las bandas de absorción, y debido a los patrones únicos que allí se encuentran, a menudo se le llama región de huella dactilar. Las bandas de absorción en la región de 4000 a 1450 cm -1 generalmente se deben a vibraciones de estiramiento de unidades diatómicas, y esto a veces se denomina región de frecuencia grupal.
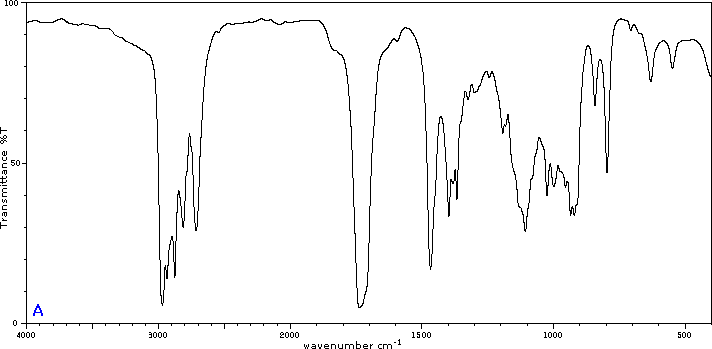
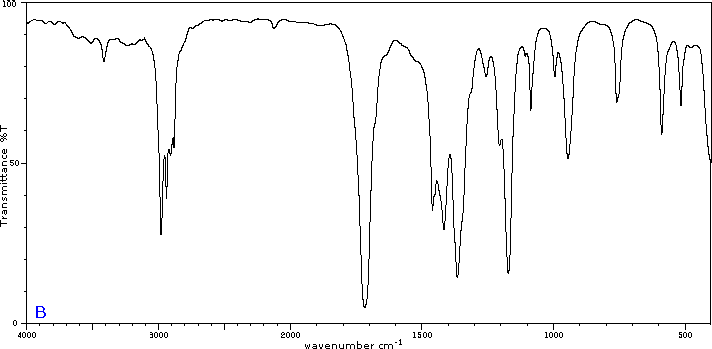
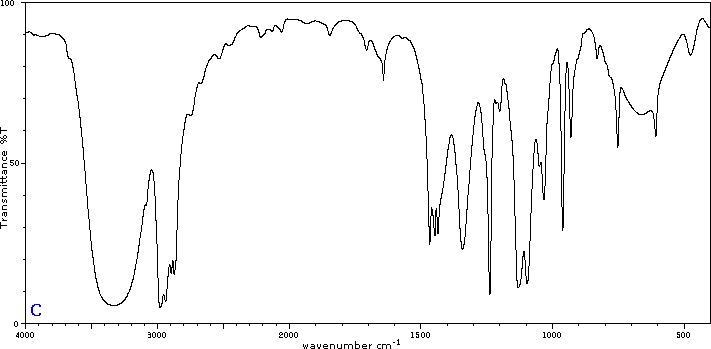
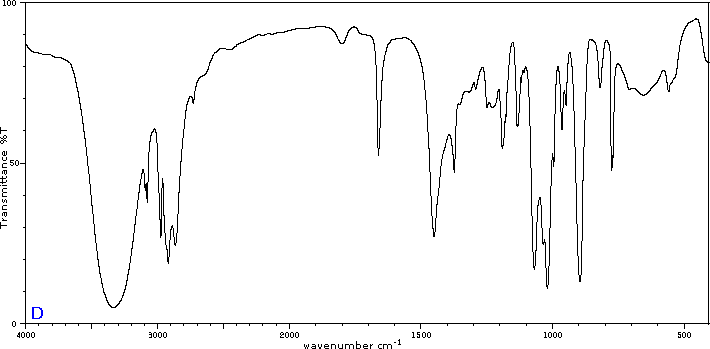
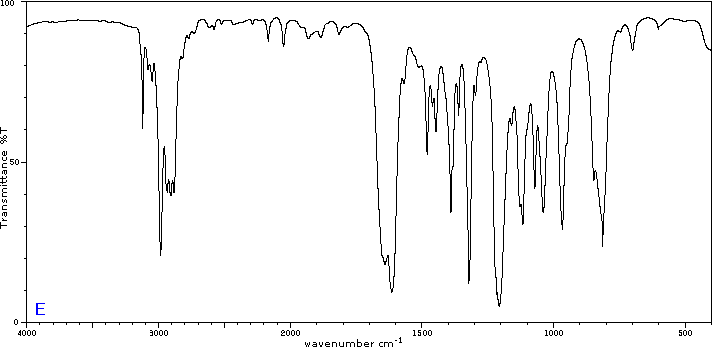
Para ilustrar la utilidad de los espectros de absorción infrarroja, a continuación se presentan ejemplos de cinco isómeros C 4H 8 O a continuación de sus correspondientes fórmulas estructurales. Trate de asociar cada espectro (A - E) con uno de los isómeros en la fila de arriba de él.
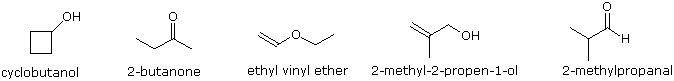
Colaboradores y Atribuciones
Dr. Dietmar Kennepohl FCIC (Professor of Chemistry, Athabasca University)
Prof. Steven Farmer (Sonoma State University)
William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry
Organic Chemistry With a Biological Emphasis by Tim Soderberg (University of Minnesota, Morris)