6.6: Espectros de RMN 1H e interpretación (Parte I)
- Page ID
- 76360

Comprender los fundamentos de la teoría de RMN nos prepara para pasar a la parte más importante y práctica de esta sección, es decir, cómo entender el espectro de RMN 1H y dilucidar la estructura de un compuesto a partir de la información del espectro de RMN 1H. Primero echemos un vistazo a un espectro de RMN 1H real.
Generalmente, la información sobre la estructura de la molécula se puede obtener a partir de cuatro aspectos de un espectro típico de RMN de 1H:
- Protonesquímicos equivalentes y no equivalentes (número total de señales)
- Turno químico
- Integración
- División de señal
6.6.1 Protones Químicos Equivalentes y No Equivalentes
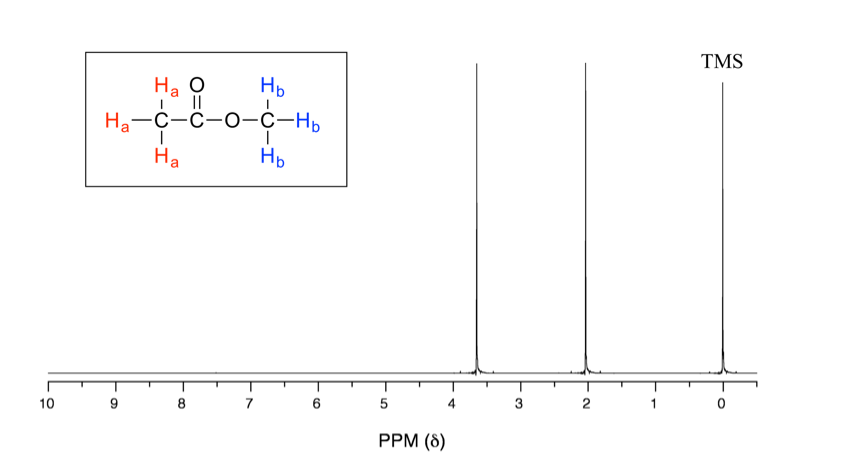
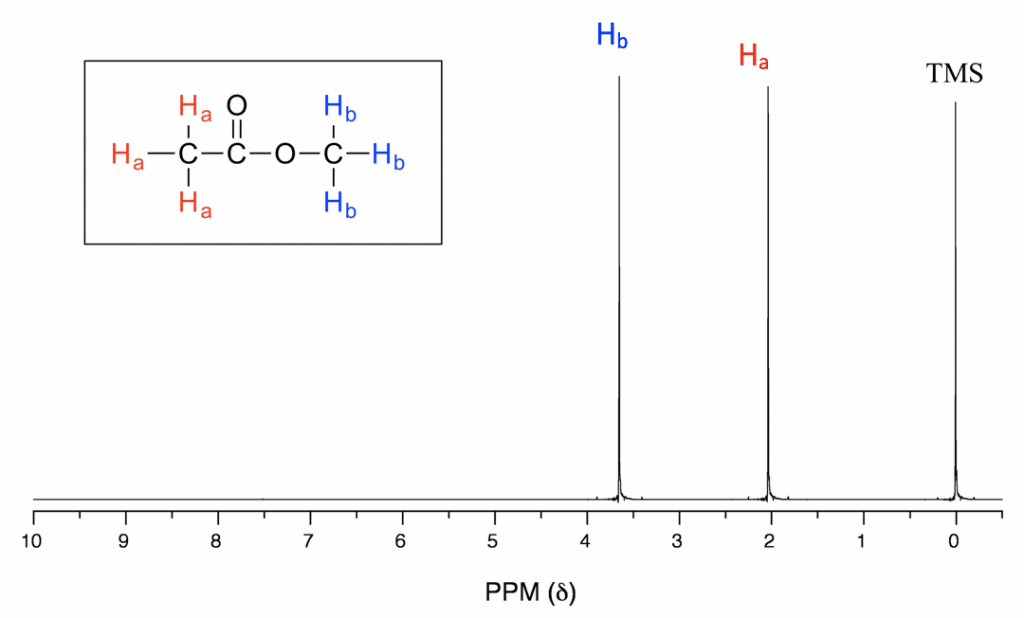
En el espectro de RMN 1H anterior del acetato de metilo (Fig. 6.6a), podemos ver que hay tres señales. El pico en el extremo derecho es para el compuesto de referencia estándar tetrametilsilano (TMS, más discusiones en la sección de desplazamiento químico 6.6.2), no para el compuesto. Por lo que el compuesto acetato de metilo muestra dos señales en espectro de RMN 1H. ¿Por qué solo dos señales para un compuesto que contiene un total de seis hidrógenos?
Esto se debe a la equivalencia química. Los seis hidrógenos totales se pueden dividir en dos grupos, los tres protones H a en el grupo metilo que se une con C=O están todos en el mismo ambiente químico, por lo tanto son equivalentes químicos. Todos los hidrógenos químicos equivalentes tienen la misma frecuencia de resonancia aplicada a un campo magnético externo, por lo que muestran solo una señal en el espectro de RMN 1H. Los tres protones H b en el grupo metilo unidos con el átomo de O también son equivalentes químicos y muestran la otra señal. Es por ello que hay un total de dos señales para el acetato de metilo compuesto.
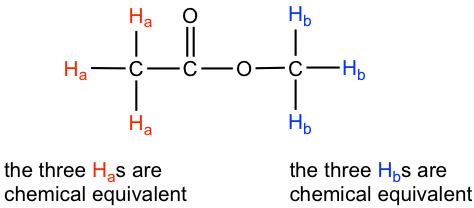
La capacidad de reconocer protones químicos equivalentes y no equivalentes en una molécula es muy importante para comprender el espectro de RMN. Para el compuesto con estructura dada, deberíamos ser capaces de predecir cuántas señales hay en el espectro de RMN 1H. Por otro lado, si el espectro de RMN 1H está disponible para un compuesto desconocido, contar el número de señales en el espectro nos indica el número de diferentes conjuntos de protones en la molécula, y esa es la información muy importante para determinar la estructura del compuesto.
Aquí pasaremos por varios ejemplos para la primera situación, es decir, predecir el número de señales en el espectro de RMN 1H con la estructura de un compuesto dada. Para ello, necesitamos contar cuántos conjuntos distintos de protones están incluidos en la molécula.
Para cada una de las siguientes moléculas, los protones químicamente equivalentes se marcan en el mismo color para facilitar la comprensión.
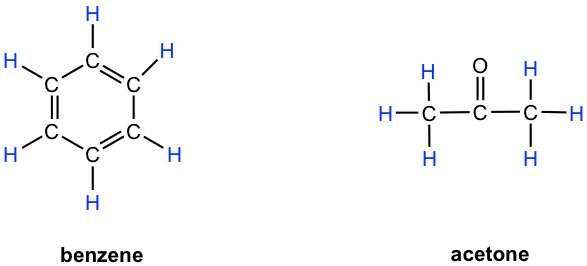
- Benceno: los seis protones son equivalentes químicos (tienen la misma unión y en el mismo ambiente químico) entre sí y tienen la misma frecuencia de resonancia en un experimento de RMN de 1H, por lo tanto, muestran solo una señal.
- Acetona: ambos grupos metilo (dos CH 3) unidos con enlace C=O, por lo que están en el mismo ambiente químico, y como resultado los seis protones son equivalentes químicos que muestran una sola señal.
Notas: Como probablemente ya se dio cuenta, la equivalencia química o no equivalencia en RMN está estrechamente relacionada con la simetría. Los protones que son simétricos entre sí por cierto plano de simetría son equivalentes químicos.
Las moléculas de la siguiente figura contienen más conjuntos de protones químicamente equivalentes.
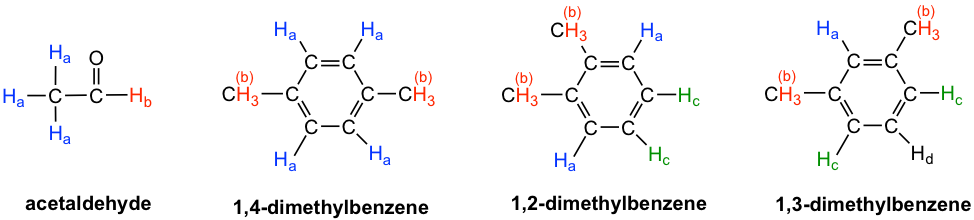
- Acetaldehído: Los tres protones H a en el grupo metilo son equivalentes químicos, y todos se enlazan a un carbono hibridado sp 3; pero son diferentes al protón H b que está unido a un carbono carbonílico hibridado sp 2. Dos señales totales en espectro de RMN 1H.
- 1,4-dimetilbenceno: los cuatro protones aromáticos son químicamente equivalentes debido a la simetría. Los dos grupos metilo son equivalentes entre sí también. Dos señales totales en espectro de RMN 1H.
- 1,2-dimetilbenceno: ambos protones H a están adyacentes a un sustituyente metilo, mientras que ambos protones H c están a dos carbonos de distancia. Entonces los cuatro protones aromáticos se dividen en dos conjuntos. Ambos grupos metilo están en el mismo enlace y simétricos entre sí, son equivalentes. Tres señales totales en espectro de RMN 1H.
- 1,3-dimetilbenceno: H b está situado entre dos grupos metilo, los dos protones H c están a un carbono de distancia de un grupo metilo y H d está a dos carbonos de un grupo metilo. Por lo tanto, los cuatro protones aromáticos se pueden dividir en tres conjuntos. Los dos grupos metilo son equivalentes. Cuatro señales totales en espectro de RMN 1H.
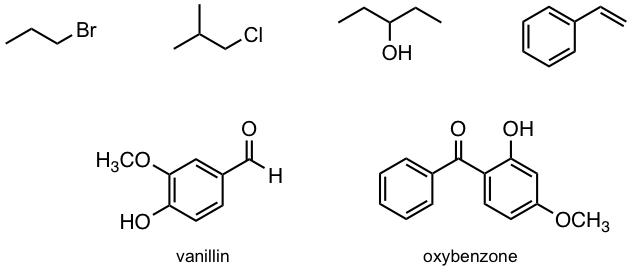
Ejercicios 6.1
¿Cuántas señales de RMN de 1H predecirías para cada una de las siguientes moléculas?
6.6.2 Desplazamiento Químico
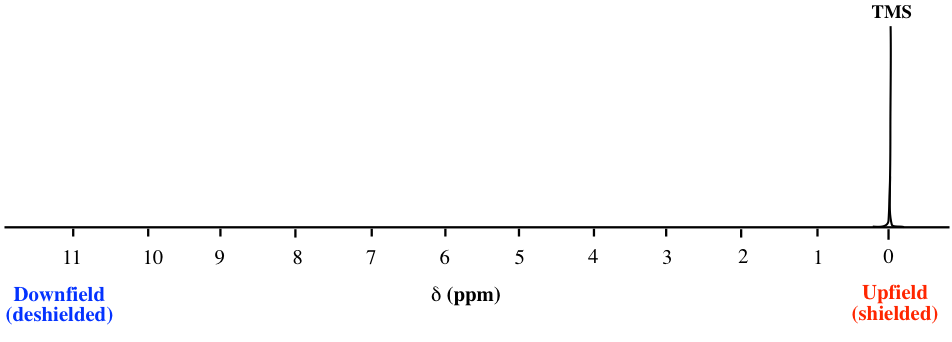
Como se observa en el espectro de RMN 1H del acetato de metilo (Fig. 6.6a), las unidades del eje x del espectro de RMN están en ppm (no en Hz como esperaríamos para la frecuencia), y las dos señales se encuentran en diferentes posiciones a lo largo del eje x. Expliquemos cómo funciona eso y qué información se puede obtener.
La posición de una señal a lo largo del eje x de un espectro de RMN se denomina desplazamiento químico, o δ, de la señal. El desplazamiento químico está determinado por el ambiente electrónico estructural de los núcleos que producen esa señal. Los protones en diferentes ambientes químicos (no equivalentes) muestran señales en diferentes desplazamientos químicos. La dirección de la escala de desplazamiento químico en el eje x es opuesta a lo que estamos familiarizados, es decir, el valor menor está en el lado derecho, y el valor mayor está en el lado izquierdo (Fig. 6.6b).
- Los valores de desplazamiento químico más pequeños (δ) corresponden con una frecuencia de resonancia más baja;
- Los valores mayores de desplazamiento químico (δ) corresponden con mayor frecuencia de resonancia.
Por convención, el lado derecho de un espectro de RMN con valores de desplazamiento químico más pequeños se llama campo arriba, y la dirección izquierda se llama campo abajo (Fig. 6.6b).
Para los protones que están apantallados, debido al Blocal causado por los electrones circulantes, el campo magnético que experimenta el protón, Beff, es menor que el campo externo aplicado, Bo, por lo que los protones resonan a menor frecuencia y tienen valores de desplazamiento químico menores.
- Los protones blindados tienen menor frecuencia de resonancia y valores de desplazamiento químico (d) más pequeños;
- Los protones deshirados tienen mayor frecuencia de resonancia y mayores valores de desplazamiento químico (d).
En el espectro de RMN 1H, la absorción de los protones de TMS (tetrametilsilano) se define como “cero” en la escala de desplazamiento químico (δ), y la absorción de otros protones se reporta como desplazamiento relativo en comparación con la del TMS.
Se eligió TMS como compuesto de referencia y se definió como “cero” por varias razones. Dado que el silicio es menos electronegativo que el carbono, los hidrógenos del TMS se encuentran en un ambiente de alta densidad de electrones, por lo tanto están altamente protegidos con muy baja frecuencia de resonancia y rara vez interfieren con las señales de otros compuestos. También hay doce hidrógenos equivalentes en TMS que muestran una sola señal, por lo que la señal es bastante fuerte incluso con muy poca cantidad de TMS. TMS también es bastante inerte y fácil de eliminar con el punto de ebullición de 27 ºC. Se utilizó una pequeña cantidad de TMS para ser agregado en la muestra como patrón interno para la medición de RMN, y luego se eliminó por evaporación. Sin embargo, para el espectrómetro de RMN contemporáneo (incluido el RMN de sobremesa), ya no es necesario agregar TMS ya que la computadora puede calibrar el desplazamiento químico electrónicamente en función de las frecuencias de resonancia del disolvente utilizado.
La unidad de desplazamiento químico (δ) es ppm. La etiqueta 'ppm' significa 'partes por millón'. El desplazamiento químico relativo al TMS en ppm se define como la siguiente fórmula.
La razón para usar un valor relativo de desplazamiento químico en ppm, en lugar de la frecuencia de resonancia real en Hz es que cada instrumento de RMN tendrá una intensidad de campo magnético diferente, por lo que el valor real de las frecuencias de resonancia expresadas en Hz será diferente en diferentes instrumentos; recuerde que ΔE para la transición magnética de un núcleo depende de la intensidad del campo magnético aplicado externamente B o. Sin embargo, el desplazamiento químico expresado en ppm siempre será el mismo ya sea medido con un instrumento que opere a 400 MHz o 60 MHz. En la RMN 1H del acetato de metilo, las dos señales están a 2.0 y 3.6 ppm representa que los dos conjuntos de protones en acetato de metilo tienen frecuencias de resonancia alrededor de 2.0 y 3.6 partes por millón mayores que la frecuencia de resonancia de los protones TMS. Si, por ejemplo, el espectro es medido por el espectrómetro de RMN de 400 MHz, entonces el desplazamiento químico en Hz será de 800 Hz y 1440 Hz respectivamente.
La mayoría de los protones en los compuestos orgánicos tienen valores de desplazamiento químico entre 0 y 12 ppm en relación con el TMS, aunque ocasionalmente se observan valores por debajo de 0 ppm y superiores a 12 ppm. El valor de desplazamiento químico de los hidrógenos en cierto ambiente estructural, o grupos funcionales orgánicos comunes, se enumeran en la tabla (Fig. 6.6c) y la tabla (Cuadro 6.2) a continuación.
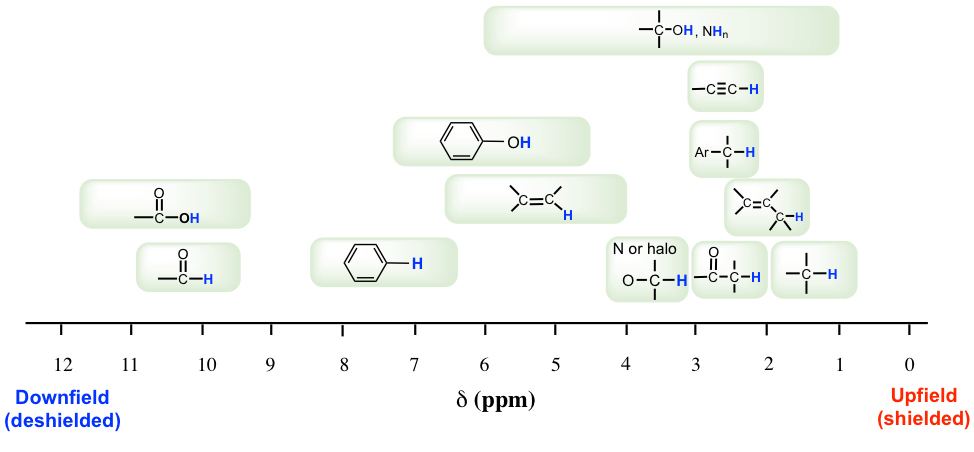
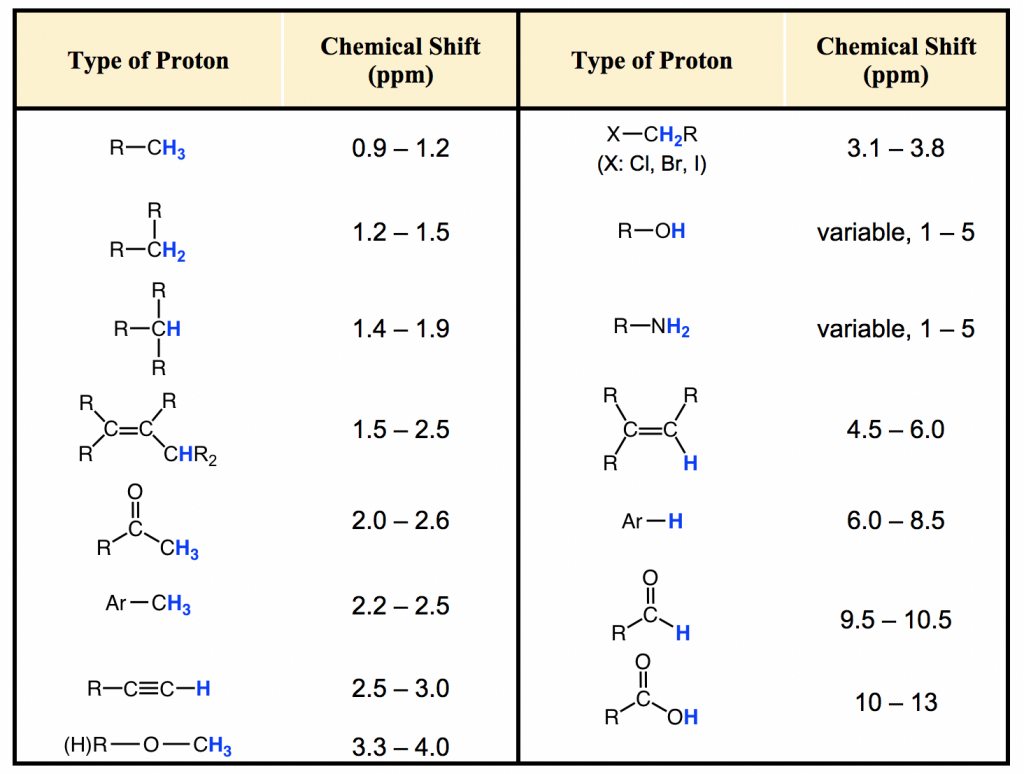
La importancia de la información de desplazamiento químico es que da pistas críticas sobre las estructuras moleculares. Varios aspectos destacados aquí:
- Por lo general, los hidrógenos en enlace C-H, sin ningún otro grupo funcional cercano, están en el rango de 1-2 ppm;
- Para el hidrógeno en el enlace C-H junto al doble enlace, como el enlace C=C o C=O, la señal va campo abajo a 2-2.5 ppm;
- Con átomos electronegativos conectados en el carbono, como O-C-H, los hidrógenos se deshielding y el desplazamiento químico se mueve más abajo a 3-4 ppm;
- Los hidrógenos unidos directamente al carbono de doble enlace tienen el desplazamiento químico alrededor de las 4.5-6 pm;
- Los hidrógenos aromáticos (H en el anillo de benceno) muestran un desplazamiento químico alrededor de 7 ppm;
- El desplazamiento químico de los hidrógenos en el grupo OH (alcohol) o NH (amina) varía en un rango bastante grande, de 1-5 ppm;
- El hidrógeno en el grupo aldehído (-CHO) y ácido carboxílico (COOH) tiene el desplazamiento químico más bien campo abajo a aproximadamente 9-10 ppm y 10-12 ppm respectivamente.
Al hacer referencia a la tabla de cambios químicos (o gráfico) para un determinado compuesto, es útil tener en cuenta que el valor exacto puede variar un poco al rango dado, en ocasiones la diferencia de hasta 0.5 ppm unidad puede ocurrir depende de la estructura específica y el disolvente utilizado.
Con la información de desplazamiento químico disponible, ahora podemos asignar las señales en el espectro de RMN 1H del acetato de metilo. Según la Fig. 6.6c, los protones en el grupo CH 3 junto al enlace C=O se supone que están en el rango de 2-3 ppm, y los protones en el grupo CH 3 conectados con O directamente tienen un valor δ de aproximadamente 3-4 ppm. Entonces la señal de 2.0 ppm es para el grupo H a y la señal de 3.6 ppm es para el grupo H b.
Desplazamiento Químico de Protones Cerca de electrones π — Efecto Anisotropía
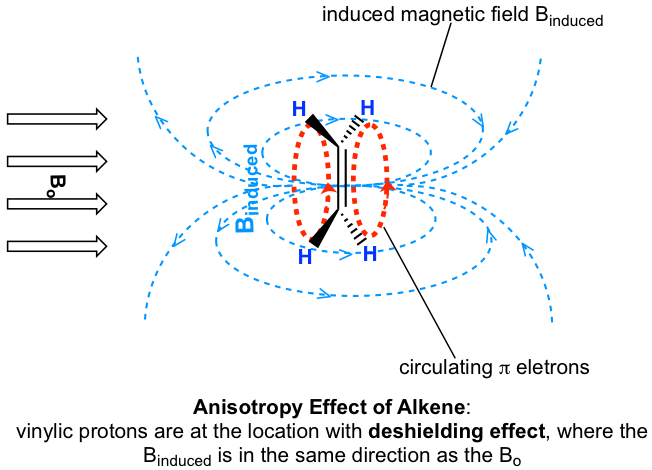
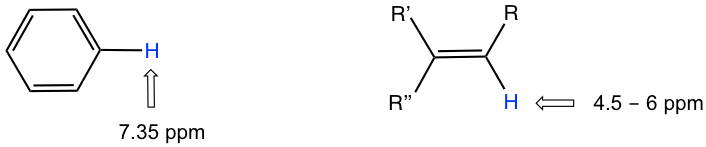
Los valores de desplazamiento químico de los protones aromáticos y los protones vinílicos (aquellos directamente unidos a un carbono alqueno) resuenan mucho más abajo (mayor frecuencia, mayor desplazamiento químico) de lo que puede explicarse simplemente por el efecto deshielding de los átomos electronegativos cercanos. Estos cambios químicos son el resultado del efecto anisotropía.
Investiguemos primero los protones aromáticos. En el anillo de benceno (y muchas otras estructuras aromáticas), el total de seis electrones π forman un gran enlace π deslocalizado alrededor del anillo (más discusiones en Organic II). Cuando la molécula se expone al campo magnético externo B o, estos electrones π comienzan a circular en una corriente de anillo y generando su propio campo magnético inducido B inducido. Si se produce blindaje o deshielding depende de la ubicación de los protones en el campo magnético inducido, y esto se llama efecto anisotropía (significa “no uniformidad”). Esto se puede ilustrar específicamente en la siguiente figura comparando entre los puntos A y B.
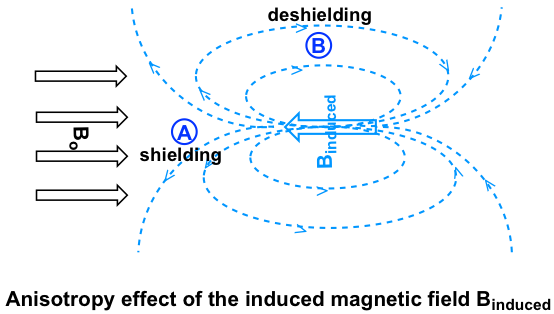
Si un protón está en el punto A, siente el campo magnético inducido apuntando a la dirección opuesta de B o, por lo que el protón experimenta un efecto de blindaje. Para el protón en el punto B, sin embargo, siente el campo magnético inducido en la misma dirección que B o, por lo que el protón experimenta un efecto de deshielding.
Los protones en el anillo de benceno están en la posición equivalente al 'punto B', es decir, que la corriente inducida en esta región del espacio está orientada en la misma dirección que B 0, por lo que se suma a B 0 y da como resultado un efecto de deshilelding y el benceno resonancia de protones a mayor frecuencia y tienen mayores desplazamientos químicos.
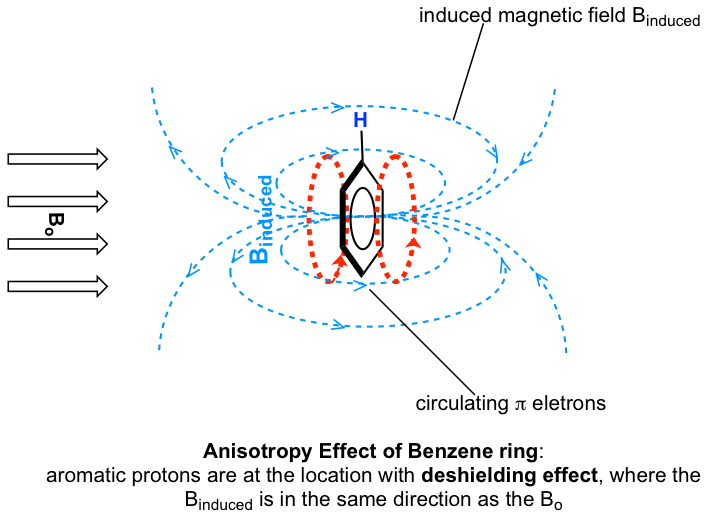
Como resultado, debido a la anisotropía del campo inducido generado por los electrones π circulantes, los protones benceno son altamente deshirados. Su desplazamiento químico está muy abajo, en el rango de 6.5—8.5 ppm.
La anisotropía también es responsable de los desplazamientos químicos en el campo bajo (alta frecuencia) de los protones vinílicos (4—6.5 ppm) y los protones aldehídos (9.5—11 ppm). Los electrones π en estos grupos también circulan de tal manera que generan un campo magnético inducido que se suma al campo externo B o en los puntos ocupados por los protones. Los protones de ácido carboxílico están aún más abajo (9.5—12 ppm) debido a la influencia combinada del átomo de oxígeno electronegativo y el enlace π cercano.