
4.4: Talasemia
- Page ID
- 123164

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)https://pressbooks.library.ualberta.ca/mlsci/?p=386
Las imágenes muestran frotis de sangre periférica de talasemia con glóbulos rojos hipocrómicos, microcíticos y poiquilocitosis. De Colección MLS, Universidad de Alberta.
Imagen 1:50x inmersión en aceite. https://doi.org/10.7939/R3DR2PQ4J
Imagen 2:50x inmersión en aceite. https://doi.org/10.7939/R3V698T05
Imagen 3:50x inmersión en aceite. https://doi.org/10.7939/R3HD7P773
Las talasemias se clasifican como un grupo de trastornos genéticos de la hemoglobina donde se ve afectada la producción de cadenas α y β globina. Esto se considera un trastorno cuantitativo de la hemoglobina y se categoriza por la cadena de globina afectada (alfa o beta), y como mayor o menor dependiendo de la gravedad de la enfermedad. 1,2
Alfa-talasemia:
Causa (s):
Los genes de la cadena α globina se localizan en el cromosoma 16 y normalmente hay cuatro genes en total (αα/αα), dos heredados de cada progenitor. La α-talasemia resulta cuando hay una deleción en cualquier número del gen α globina. La gravedad de la anemia y la cantidad de producción de la cadena de α globina depende del número de genes que se eliminan. 3
Transportador Silencioso α-Talasemia (αα/α-): 1,2
Ocurre cuando se elimina un gen α. Todavía hay una producción adecuada de α para asegurar la síntesis normal de hemoglobina. El paciente es asintomático y la mutación es benigna.
En los recién nacidos, hay un exceso de producción de cadenas de γ globina. Estas cadenas de γ globina tienden a formar tetrámeros y dan como resultado Hemoglobin Barts (Hb Barts). Hb Barts tiene una alta afinidad de oxígeno y es ineficiente para el suministro de oxígeno a los tejidos del feto en desarrollo. En el estado de portador silencioso, sólo se produce una pequeña cantidad de Hb Barts.
α-Talasemia Menor (αα/—) o (α-/α-): 1,2
Ocurre cuando se eliminan dos genes α. Ahora hay una reducción del 50% en la producción normal de la cadena de α globina.
En adultos, el aumento de la producción de glóbulos rojos es capaz de compensar la disminución en la producción de la cadena α, y la producción de la cadena α y β globina está equilibrada. Los pacientes son asintomáticos y cualquier anemia presente es leve.
Hay entre 5-15% de hemoglobina Barts presente al nacer, pero esto disminuye una vez que la producción de la cadena β globina toma el control y la producción de la cadena de γ globina disminuye. En adultos, la producción de la cadena de globina es equilibrada, por lo que no se forma Hemoglobina H.
Enfermedad de la hemoglobina H (α-/—): 1,2

- Imagen de un frotis de sangre periférica de la Enfermedad de Hemoglobina H que muestra marcada poiquilocitosis (células en forma de lágrima, esquistocitos, células diana y eliptocitos). Inmersión en aceite de 50x. De la Colección MLS, Universidad de Alberta, https://doi.org/10.7939/R30P0X613
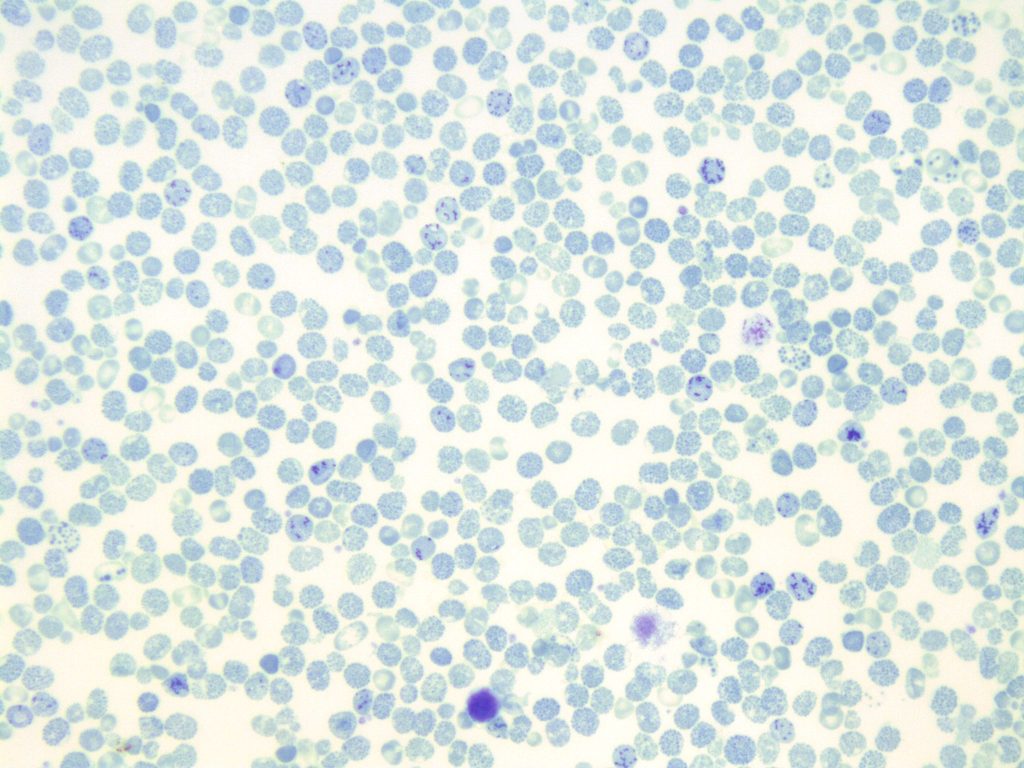
- Frotis de sangre periférica con la Enfermedad de Hemoglobina H con tinción supravital. Se observan inclusiones de hemoglobina H en los glóbulos rojos (como pelota de golf). Inmersión en aceite 50x. De la Colección MLS, Universidad de Alberta, https://doi.org/10.7939/R3BV7BB0V
Ocurre cuando se borran tres genes α. La producción de genes α se reduce significativamente (75% de reducción) provocando un mayor desequilibrio entre el número de cadenas de globina α y β que se producen. Los pacientes presentan una anemia hemolítica crónica que varía de leve a moderada. Los pacientes son transfusionales independientes.
El exceso de cadenas de globina β forman tetrámeros conocidos como Hemoglobina H (Hb H). La Hb H es inestable y a menudo precipita dentro de los glóbulos rojos dando como resultado anemia hemolítica. Se incrementa la producción de Hb Barts al nacer.
Hidropesía fetalis/ α-talasemia mayor (—/—): 1
Ocurre cuando se eliminan los cuatro genes α (sin producción de cadena de α globina).
Debido a que no se produce una cantidad sustentable de cadenas de α globina, este estado generalmente se considera incompatible con la vida. El exceso de cadenas de γ globina da como resultado la formación de Hb Barts. Debido a su alta afinidad por el oxígeno, no es capaz de transportar oxígeno de manera eficiente a los tejidos del feto en desarrollo. La marcada hipoxia tisular suele dar como resultado la muerte fetal en el útero o poco después del nacimiento.
Cuadro 1. Hallazgos de laboratorio de α-talasemias 1
α-Talasemia Estado | CBC y RETIC | PBS | BM | Contenido de Hemoglobina |
Transportador Silencioso α-Talasemia (αα/α-) | Hb: Normal RBC: Normal MCV/MCH/MCHC: Normal RDW: Normal RETIC: Normal |
| Normal | Hb A: Normal Hb Barts: 1-3% al nacer Hb H: 0% |
α-Talasemia Menor (αα/—) o (α-/α-) | Hb: Normal a Disminución RBC: Normal a Aumentado MCV/MCH/MCHC: Disminuido RDW: Normal RETIC: Elevado |
| Hiperplasia Eritroide | Hb A: Disminución leve Hb Barts: 5-15% al nacer Hb H: 0% |
Enfermedad de la Hemoglobina H (α-/—) | Hb: Disminución RBC: Incrementado MCV/MCH/MCHC: Disminuido RDW: Normal a Aumentada RETIC: Elevado |
| Hiperplasia Eritroide | Hb A: Disminución Hb Barts: 10-40% al nacer, huellas en adultos Hb H: 1-40% en adultos |
Fetálicos de hidropes/ α-Talasemia Mayor (—/—) | Hb: Disminución RBC: Disminuido MCV/MCH/MCHC: Disminuido RDW: Incrementado RETIC: Elevado |
| Hiperplasia Eritroide | Hb A: 0% Hb Barts: 80-90% Hb H: No formado Ausentes: Hb A, Hb A 2, Hb F |
Beta-talasemias
Causa (s):
Los genes de la cadena β globina se localizan en el cromosoma 11 y normalmente hay dos genes en total (β/β) uno heredado de cada progenitor. La β-talasemia generalmente se debe a mutaciones puntuales en los genes de β globina. Estas mutaciones puntuales hacen que la producción de cadenas de β globina se reduzca (β +) o se suprima completamente (β 0). 3
Transportador Silencioso β-Talasemia (β Silencioso/β): 2
La mutación de los genes de la cadena β globina no da como resultado ningún hallazgo hematológico anormal y la producción de la cadena β globina es normal o casi normal.
β-Talasemia Menor (β 0 /β o β + /β): 1,2
Un gen de la cadena β globina está mutado mientras que el otro gen de la cadena de globina β es normal. El paciente es capaz de producir suficientemente suficientes cadenas de β globina para mantener la oxigenación normal y la vida útil de los glóbulos rojos.
Los pacientes son asintomáticos y presentan anemia leve que puede empeorar en condiciones de estrés.
β-Talasemia Intermedia (β + /β Silencioso o β 0 /β Silencioso o β Silencioso /β Silencioso): 2
Las mutaciones en los genes β resultan en una producción reducida de la cadena de globina β. Los síntomas clínicos son variables y más graves que la β-talasemia menor, aunque los pacientes no requieren transfusiones para sobrevivir.
β-talasemia mayor (β + /β + o β + /β 0 o β 0 /β 0): 1,2
Las mutaciones en ambos genes β resultan en una producción severamente disminuida o ausente de cadenas de globina β. El exceso de cadenas de α globina son incapaces de formar tetrámeros que conducen a su precipitación y acumulación en el glóbulo rojo. Esto daña la célula y resulta en una anemia hemolítica crónica y severa.
Los pacientes requieren transfusiones regulares.
Cuadro 2. Hallazgos de laboratorio de β-talasemias 2
| β-Talasemia Estado | CBC y RETIC | PBS | BM | Contenido de Hemoglobina |
| Transportador Silencioso β-Talasemia (β Silencioso /β) | Hb: Normal RBC: Normal MCV/MCH/MCHC: Normal RDW: Normal RETIC: Normal | N/A | N/A | Hb A: Normal Hb A 2: Normal Hb F: Normal |
| β-Talasemia Menor (β 0 /β o β + /β) | Hb: Disminución RBC: Normal a Aumentado MCV/MCH/MCHC: Disminuido RDW: Normal RETIC: Normal a Aumentada |
| Hiperplasia Eritroide | Hb A: 92-95% Hb A 2: 3.5-7.0% Hb F: 1-5% |
| β-Talasemia Intermedia (β + /β Silencioso o β 0 /β Silencioso o β Silencioso /β Silencioso) | Hb: Disminución RBC: Incrementado MCV/MCH/MCHC: Disminuido RDW: Normal RETIC: Incrementado |
| Hiperplasia Eritroide | Hb A: Disminución Hb A 2: Incrementado Hb F: Incrementado |
| β-Talasemia Mayor (β + /β + o β + /β 0 o β 0 /β 0) | Hb: Disminución RBC: Incrementado MCV/MCH/MCHC: Disminuido RDW: Normal a Aumentada RETIC: Incrementado |
| Hiperplasia Eritroide (Eritropoyesis ineficaz) | Hb A: Ausente o disminuida Hb A 2: Variable Hb F: 70-90% |
Otras pruebas de laboratorio para evaluar la talasemia: 2
Estudios del Hierro (Los estudios de Thalasemia Iron se muestran en el siguiente capítulo)
Electroforesis de hemoglobina
Cromatografía líquida de alto rendimiento (HPLC)
Pruebas moleculares para mutaciones/deleciones genéticas
Notas:
Los resultados anteriores son solo hallazgos típicos, los cuales pueden ser alterados dependiendo de la variación individual, el tratamiento dado (como las transfusiones de glóbulos rojos) y el subtipo genético.
El cuadro de frotis de sangre periférica para las formas menores de Talasemia se ve muy similar al de la Anemia por Deficiencia de Hierro. La diferencia entre las dos condiciones se puede distinguir comparando los resultados del estudio de hierro, así como hallazgos específicos de CBC (RDW, recuento de glóbulos rojos) y hallazgos de frotis periféricos (inclusiones, poiquilocitosis). 2
Referencias:
1. Randolph TR. Talasemia. En: Hematología de laboratorio clínico. 3ª ed. Nueva Jersey: Pearson; 2015. p. 251-276.
2. Keohane Talasemias. En: Aplicaciones y principios clínicos de hematología de Rodak. 5ta ed. San Luis, Misuri: Saunders; 2015. p. 454-74.
3. Chonat S, Quinn CT. Estándares actuales de atención y resultados a largo plazo para talasemia y anemia falciforme. Adv Exp Med Biol [Internet]. 2017 [citado 2018 Jun 5]; 1013:59 —87. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC5720159/