
8.5: Transcriptómica
- Page ID
- 53237
Considere una matriz que contenga todas las secuencias génicas conocidas en un genoma. Para hacer tal matriz para su análisis, se necesitaría hacer copias de cada gen, ya sea por síntesis química o usando la reacción en cadena de la polimerasa. Las cadenas de los ADN resultantes se separarían entonces para obtener secuencias monocatenarias que podrían unirse al chip. Cada caja de la cuadrícula contendría la secuencia de un gen. Con esta cuadrícula, se podría analizar el transcriptoma - todos los ARNm se hacen en células seleccionadas en un momento dado. Para un análisis simple, se podría tomar un tejido (digamos hígado) y extraer de él todos los ARNm. Esta población de ARNm representa todos los genes que se estaban expresando en las células hepáticas en el momento en que se extrajo el ARN. Estos ARN deberían poder hibridarse (pares de bases) con sus genes correspondientes en la micromatriz. Los genes que no se expresaban no tendrían ARNm para unirse a sus genes correspondientes en la cuadrícula.
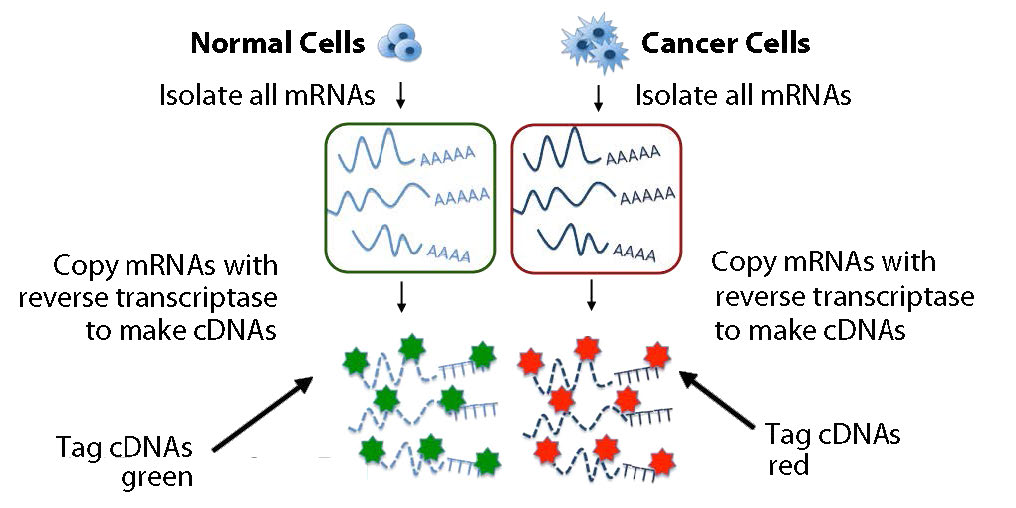
Figura 8.25 - Copia y marcaje del transcriptoma. Imagen de Taralyn Tan
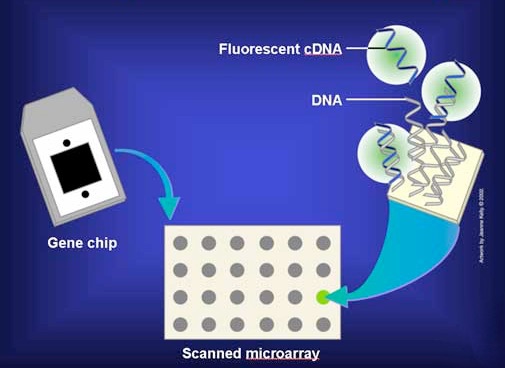
En la práctica, los ARNm no se utilizan directamente, sino que se copian en copias de ADN monocatenario llamadas ADNc. Los ADNc se etiquetan con un colorante fluorescente y se agregan a la micromatriz en condiciones que permiten el emparejamiento de bases para que los ADNc puedan encontrar y emparejar bases con secuencias complementarias en la matriz (Figura 8.26). Luego se lava la matriz para eliminar los ADNc no hibridados. La presencia/ausencia/abundancia de cada ARNm se determina entonces fácilmente midiendo la cantidad de colorante en cada caja de la cuadrícula.
Figura 8.26 - Agregar ADNc marcados a la placa de micromatrices. Imagen de Taralyn Tan
En la Figura 8.27, un ADNc fluorescente se ha unido al punto del extremo derecho en la tercera fila de la cuadrícula. Esto significa que la secuencia del ADNc era complementaria a la secuencia de la secuencia génica inmovilizada en ese punto. Debido a que se conoce la identidad de los genes en cada posición de la cuadrícula, entonces sabemos que la muestra contenía ARNm que correspondía a ese gen en particular. Es decir, ese gen se estaba expresando en las células de las que se obtuvieron los ARNm.
Se podría realizar un análisis más potente con dos conjuntos de ARNm simultáneamente. Un conjunto de ADNc podría provenir de un tejido canceroso y el otro de un tejido no canceroso, por ejemplo. Los ADNc derivados de cada muestra están marcados con un color diferente (digamos verde para normal y rojo para canceroso) (Figura 8.25). Los ADNc se mezclan y luego se agregan a la matriz y se permite nuevamente que las secuencias complementarias formen dúplexes (Figura 8.27).
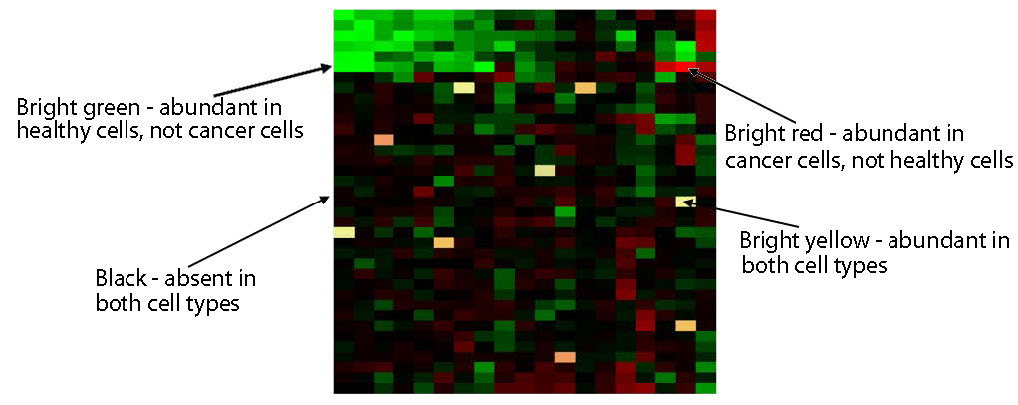
Figura 8.28 - Análisis de micromatrices que compara la expresión génica en células normales y cancerosas. Wikipedia
Los ADNc no hibridados se eliminan por lavado y luego se analiza la placa. Las cajas rojas corresponden a un ARNm presente en el tejido canceroso, pero no en el tejido no canceroso. Las cajas verdes corresponden a un ARNm presente en el tejido no canceroso, pero no en el tejido canceroso. El amarillo correspondería a los ARNm presentes en igual abundancia en los dos tejidos (Figura 8.28). La intensidad de cada mancha también da información sobre las cantidades relativas de cada ARNm en cada tejido.

Figura 8.29 - Secuenciador automatizado de alto rendimiento. Wikipedia
El mismo principio utilizado para las micromatrices de ácidos nucleicos se puede adaptar para analizar otras moléculas. Por ejemplo, los polipéptidos podrían unirse al portaobjetos de vidrio en lugar de ADN para crear un chip de proteína. Los chips de proteína son útiles para estudiar las interacciones de proteínas con otras moléculas así como para el diagnóstico.
Técnica RNA-seq
Al igual que las micromatrices, un método más nuevo llamado RNA-seq, es una herramienta para detectar y cuantificar simultáneamente todos los transcritos en una muestra dada. Este método se basa en tecnologías de secuenciación recientemente desarrolladas llamadas secuenciación de próxima generación, o secuenciación profunda. Estas técnicas permiten la secuenciación rápida y paralela de millones de fragmentos de ADN y, por lo tanto, pueden usarse no solo para ADN genómico, sino también para secuenciar todos los ARN transcritos inversamente de una muestra dada.
Para determinar todos los genes codificantes de proteínas que se expresaban en un conjunto particular de células bajo condiciones fisiológicas específicas, primero se extraería todo el ARNm y se transcribiría de forma inversa en ADNc. Este paso es similar a la preparación de muestras para microarrays. Sin embargo, en este punto, los ADNc están fragmentados en trozos más pequeños, y tienen pequeños adaptadores de secuenciación unidos en cada extremo. Los fragmentos se someten luego a secuenciación de alto rendimiento, para obtener secuencias cortas de todos los fragmentos. Estos datos se alinean contra la secuencia del genoma y se utilizan para medir el nivel de expresión de diferentes genes. RNA-seq ofrece algunas ventajas sobre las micromatrices. Con microarrays, un ARN solo puede detectarse si la secuencia génica correspondiente está presente en la cuadrícula. En RNA-seq cada ARN presente en la muestra es secuenciado, por lo que la detección de ARN no está limitada por las sondas en un chip. ARN-seq es más sensible que las micromatrices y ofrece un rango mucho mayor en el que la expresión génica se puede medir con precisión.