
10.7: Aproximación probabilística al problema del plegamiento del ARN
- Page ID
- 54933

Secuencia codificante de ARN dentro del genoma Encontrar secuencias codificantes de ARN dentro del genoma es un problema muy difícil. Sin embargo hay formas de hacerlo. Una forma es combinar la información de estabilidad termodinámica, con una puntuación RNAold normalizada y luego podemos hacer una clasificación de Máquina de Vector de Soporte (SVM), y comparar la estabilidad termodinámica de la secuencia con algunas secuencias aleatorias del mismo contenido de GC y la misma longitud y ver cuántos estándares desviaciones es la estructura dada más estable que el valor esperado.
Podemos combinarlo con la medida evolutiva y ver si el ARN está más conservado o no. Esto nos da (con relativa precisión) una idea de si la secuencia genómica en realidad está codificando un ARN.
Hemos estudiado sólo la mitad de la historia. Si bien el enfoque termodinámico es una buena manera (y la clásica) de plegar los ARN, a alguna parte de la comunidad le gusta estudiarlo desde un aspecto diferente. Asumamos por ahora que no
Secuencia codificante de ARN dentro del genoma
Encontrar secuencias codificantes de ARN dentro del genoma es un problema muy difícil. Sin embargo hay formas de hacerlo. Una forma es combinar la información de estabilidad termodinámica, con una puntuación de pliegue de ARN normalizada y luego podemos hacer una clasificación de Máquina de Vector de Soporte (SVM), y comparar la estabilidad termodinámica de la secuencia con algunas secuencias aleatorias del mismo contenido de GC y la misma longitud y ver cuántos estándares desviaciones es la estructura dada más estable que el valor esperado.
saber algo sobre la física del ARN o el factor Boltzman. En cambio, nos fijamos en el ARN como una cadena de letras para la que queremos encontrar la estructura más probable. Ya nos enteramos de los Modelos Ocultos de Markov en las conferencias anteriores. Son una buena manera de hacer predicciones sobre los estados ocultos de un sistema probabilístico. La pregunta es ¿podemos usar modelos Hidden Markov para el problema del plegamiento de ARN? La respuesta es sí.
Podemos combinarlo con la medida evolutiva y ver si el ARN está más conservado o no. Esto nos da (con relativa precisión) una idea de si la secuencia genómica en realidad está codificando un ARN.
Hemos estudiado sólo la mitad de la historia. Si bien el enfoque termodinámico es una buena manera (y la clásica) de plegar los ARN, a alguna parte de la comunidad le gusta estudiarlo desde un aspecto diferente.
Supongamos por ahora que no sabemos nada de la física del ARN o del factor Boltzman. En cambio, nos fijamos en el ARN como una cadena de letras para la que queremos encontrar la estructura más probable. Ya nos enteramos de los Modelos Ocultos de Markov en las conferencias anteriores. Son una buena manera de hacer predicciones sobre los estados ocultos de un sistema probabilístico. La pregunta es ¿podemos usar modelos Hidden Markov para el problema del plegamiento de ARN? La respuesta es sí.
Podemos representar la estructura del ARN como un conjunto de estados ocultos de puntos y corchetes (recordar la representación de paréntesis de puntos del ARN en la parte 3). Aquí hay una observación importante que hacer: las posiciones y los emparejamientos dentro del ARN no son independientes, por lo que no podemos simplemente tener un estado de corche-apertura sin ninguna consideración de los eventos que están sucediendo aguas abajo.
Por lo tanto, necesitamos extender el marco HMM para permitir correlaciones anidadas. Afortunadamente, ya existe el marco probabilístico para hacer frente a tal problema. Se le conoce como gramática estocástica libre de contexto (SCFG).
Gramática libre de contexto en pocas palabras
Tienes:
- Conjunto finito de símbolos no terminales (estados) por ejemplo {A, B, C} y símbolos terminales, por ejemplo, {a, b, c}
- Conjunto finito de reglas de producción. e.g. {A → aB, B → AC, B → aa, → ab}
- Un inicial (inicio) no terminal
Quieres encontrar una manera de llegar de un estado a otro (o a una terminal). \[A→aB→aAC→ aaaC → aaaab \nonumber\]
En un CFG estocástico, la única diferencia es que cada relación tiene una cierta probabilidad.e.g.:\[P(B → AC) = 0.25 P(B → aa) = 0.75 \nonumber\]
La evaluación filogenética se combina fácilmente con scFg, ya que existen muchos modelos probabilísticos para datos filogenéticos. Los modelos probabilísticos no se discuten en detalle en esta conferencia pero la siguiente imagen básicamente da una analogía entre los modelos estocásticos y los métodos que hemos visto hasta ahora en la clase.
- Analogías al plegado termodinámico:
— CYK ↔ Energía mínima libre (Nussinov/Zuker)— Algoritmo interior/exterior ↔ Funciones de partición (McCaskill)
- Analogías a los modelos de Hidden Markov:
— CYK Mínimo ↔ Algoritmo de Viterbi
— Algoritmo de interior/exterior ↔ Algoritmo - Dado un SCFG parametrizado (θ, Ω) y una secuencia x, el algoritmo de programación dinámica Cocke-Younger-Kasami (CYK) encuentra un árbol de análisis óptimo (probabilidad máxima)\(\hat{\pi}\):
\(\hat{\pi}\)= ArgMaxProb (π, x|θ, Ω)
- El algoritmo Inside, se utiliza para obtener la probabilidad total de la secuencia dado el modelo sumado sobre todos los árboles de análisis,\[\text{Prob}(x|Θ, Ω) = \sum \text{Prob}(x, π|Θ, Ω)\]
Aplicación de SCFG
• Predicción de la estructura secundaria de consenso: Pfold — First Phylos-SCFG
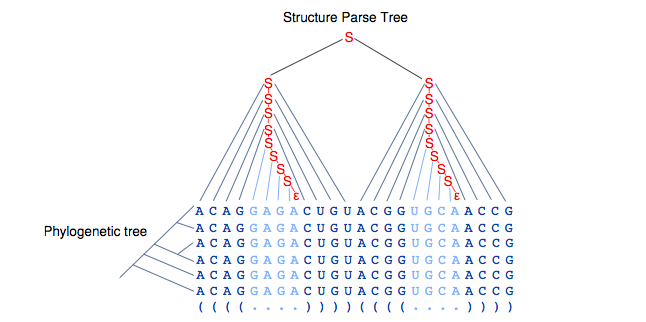
• Identificación génica de ARN estructural: EvoFold
10.8
— Utiliza gramática Pfold
— Dos modelos competidores:
∗ Modelo no estructural con todas las columnas tratadas como evolucionando independientemente
∗ Modelo estructural con columnas dependientes e independientes
— Parametrización sofisticada