
12.4: Tema práctico- RNaseQ
- Page ID
- 54490

RNA-seq es un método que utiliza tecnología de secuenciación de próxima generación para secuenciar ADNc, lo que nos permite obtener información sobre el contenido del ARN. Los dos principales problemas que aborda ARN-seq son (1) descubrir nuevos genes como las isoformas de empalme de genes previamente descubiertos y (2) descubrir los niveles de expresión de genes y transcritos a partir de los datos de secuenciación. Además, RNA-seq también está comenzando a reemplazar muchas técnicas de secuenciación tradicionales, lo que permite a los laboratorios realizar experimentos de manera más eficiente.
Cómo funciona
La máquina RNA-seq agarra un transcrito y lo rompe en diferentes fragmentos, donde los fragmentos se distribuyen normalmente. Con la velocidad con la que el RNA-seq puede secuenciar estos fragmentos de transcripción (o lecturas), hay un número abundante de lecturas que nos permiten extraer niveles de expresión. La idea básica detrás de este método se basa en el hecho de que cuanto más abundante sea una transcripción, más fragmentos secuenciaremos a partir de ella.
Las herramientas utilizadas para analizar los datos de RNA-seq se conocen colectivamente como las “Herramientas de Esmoquin”
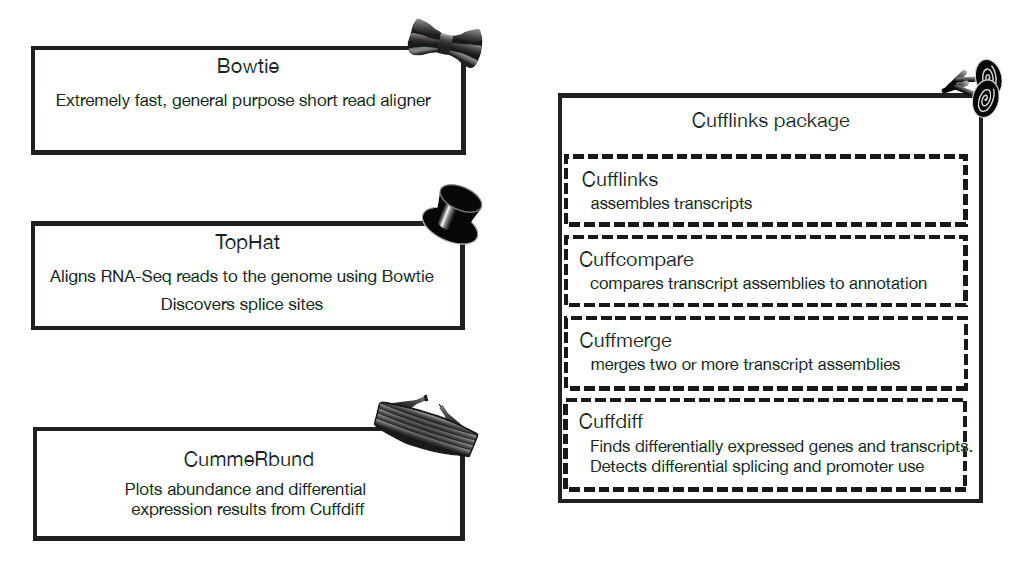
Figura 12.1: Herramientas de esmoquin
Alineación de lecturas de RNA-seq con genomas y transcriptomas
Dado que RNA-seq produce tantas lecturas, el algoritmo de alineación debe tener un tiempo de ejecución rápido, aproximadamente del orden de O (n). Existen dos estrategias principales para alinear lecturas cortas, las cuales requieren que ya tengamos las transcripciones.
1. Indización de semillas espaciadas
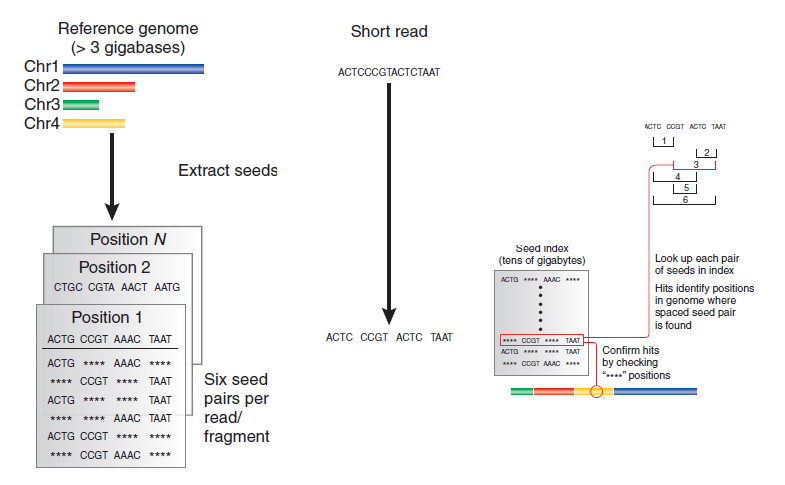
Figura 12.2: Cómo funciona la indexación de semillas espaciadas
La indexación de semillas espaciadas implica tomar cada lectura y dividirla en fragmentos, o “semillas”. Tomamos cada combinación de dos fragmentos (“pares de semillas”) y los comparamos con un índice de semillas (que tomará decenas de gigabytes de espacio) para posibles aciertos. Compara las otras semillas con el índice para asegurarte de que tenemos un acierto.
2. Indexación de Madriñas-Wheeler
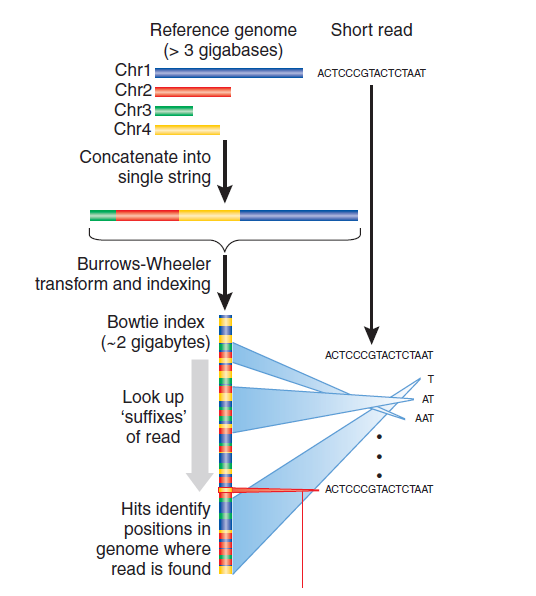
Figura 12.3: Cómo funciona la indexación de Burrows-Wheeler
La indexación de Burrows-Wheeler toma el genoma y lo revuelve de tal manera que puedes mirar el personaje leído a la vez y arrojar una gran parte del genoma como posibles posiciones de alineación muy rápidamente.
Un problema importante con estas dos estrategias de alineación de propósito general es que no tienen en cuenta grandes brechas en la alineación.
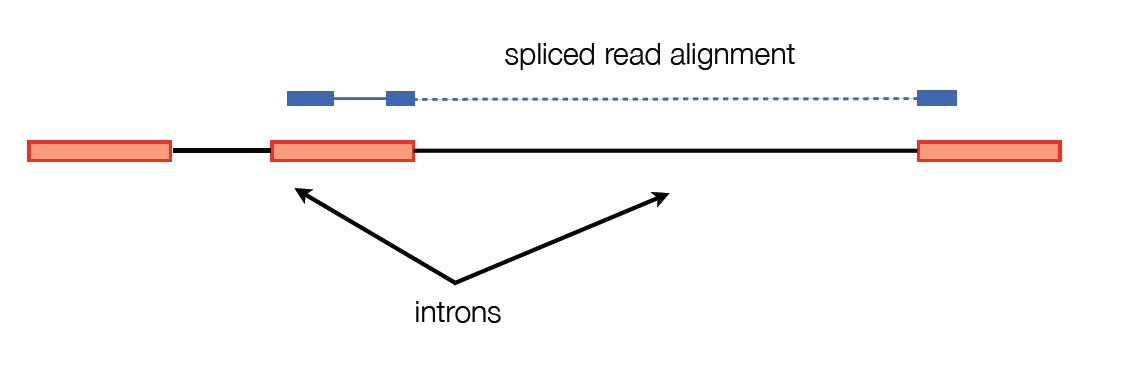
Para sortear esto, TopHat rompe las lecturas en trozos más pequeños. Estas piezas están alineadas y las lecturas con piezas que se mapean muy separadas se marcan para posibles sitios de intrones. Las piezas que no pudieron alinearse se utilizan para confirmar los sitios de empalme. Luego, las lecturas se vuelven a unir para hacer alineaciones de lectura completas.
Existen dos estrategias para ensamblar transcripciones basadas en lecturas de RNA-seq.
1. Enfoque guiado por genoma (utilizado en software como Gemelos)
La idea detrás de este enfoque es que no necesariamente sabemos si dos lecturas provienen de la misma transcripción, pero sabremos si provienen de diferentes transcripciones. El algoritmo es el siguiente: tomar las alineaciones y ponerlas en una gráfica. Agregue un borde de x → y si x está a la izquierda de y en el genoma, x e y se superponen consistentemente, e y no está contenido en x Entonces tenemos un borde de x → y si pudieran provenir de la misma transcripción.
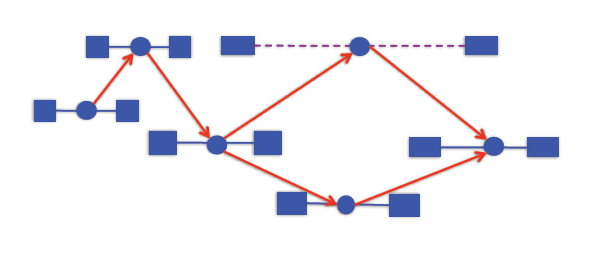
Si cruzamos esta gráfica de izquierda a derecha, obtenemos una transcripción potencial. Aplicando el teorema de Dilworth para leer órdenes parciales, podemos ver que el tamaño de la anticadena más grande en la gráfica es el número mínimo de transcripciones necesarias para explicar la alineación. Una anticadena es un conjunto de alineaciones con la propiedad de que no hay dos compatibles (es decir, podrían surgir de la misma transcripción)
2. Enfoque independiente del genoma (utilizado en software como trinity)
El enfoque independiente del genoma intenta juntar los transcritos directamente a partir de las lecturas usando métodos clásicos para el ensamblaje de lectura basado en superposición, similar a los métodos de ensamblaje del genoma.
Cálculo de la expresión de genes y transcritos
Queremos contar el número de lecturas de cada transcripción para encontrar el nivel de expresión de la transcripción. Sin embargo, dado que dividimos las transcripciones en fragmentos del mismo tamaño, nos encontramos con el problema de que las transcripciones más largas producirán naturalmente más lecturas que una transcripción más corta. Para dar cuenta de esto, calculamos los niveles de expresión en FPKM, fragmentos por kilobase por millón de fragmentos mapeados.
Función de verosimilitud para un gen
Supongamos que secuenciamos una lectura particular, la llamamos F1.
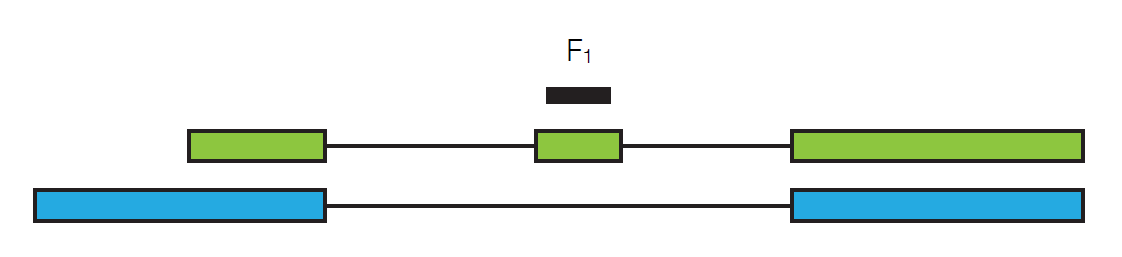
Para obtener esta lectura en particular, necesitamos elegir la transcripción particular en la que se encuentra y luego tenemos que elegir esta lectura en particular de toda la transcripción. Si\($\gamma_{\text {green }}$\) definimos que es la abundancia relativa de la transcripción verde, entonces tenemos
\[P\left(F_{1} \mid \gamma_{\text {green }}\right)=\frac{\gamma_{\text {green }}}{l_{\text {green }}}\nonumber \]
donde l verde es la longitud de la transcripción verde. Ahora supongamos que miramos una lectura diferente, F2.
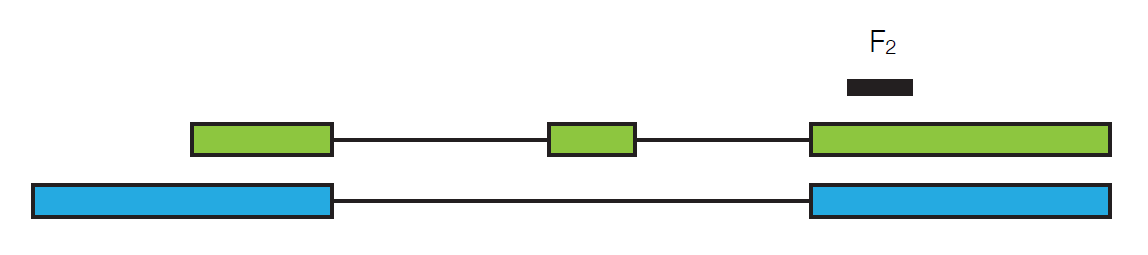
Podría haber venido ya sea de la transcripción verde de la transcripción azul, entonces:
\[P\left(F_{2} \mid \gamma\right)=\frac{\gamma_{\text {green }}}{l_{\text {green }}}+\frac{\gamma_{\text {blue }}}{l_{\text {blue }}} \nonumber \]
Podemos ver que la probabilidad de obtener tanto F1 como F2 es solo el producto de las probabilidades individuales:
\[P(F \mid \gamma)=\frac{\gamma_{\text {green }}}{l_{\text {green }}} \cdot\left(\frac{\gamma_{\text {green }}}{l_{\text {green }}}+\frac{\gamma_{\text {blue }}}{l_{\text {blue }}}\right) \nonumber \]
Definimos esto como nuestra función de verosimilitud, L (F|γ). Dada una entrada de abundancias, obtenemos una probabilidad de cuán probable es nuestra secuencia de lecturas. Entonces, a partir de un conjunto de lecturas y transcripciones, podemos construir una función de verosimilitud y calcular los valores para gamma que maximizarán esta función. Gemelos logra esto usando escalada en colina o EM en la función de verosimilitud logarítmica.
Análisis diferencial con RNA-seq
Supongamos que realizamos un análisis de RNA-seq para un gen bajo dos condiciones diferentes. ¿Cómo podemos saber si hay una diferencia significativa en los recuentos de fragmentos? Calculamos la expresión estimando el número esperado de fragmentos que provienen de cada transcripción. Para probar la significancia, necesitamos conocer la varianza de esa estimación. Modelamos la varianza como:
Var (expresión) = Variabilidad técnica + Variabilidad biológica
La variabilidad técnica, que es la variabilidad de la incertidumbre en las lecturas de mapeo, se puede modelar bien con una distribución de Poisson (ver figura a continuación). Sin embargo, el uso de Poisson para modelar la variabilidad biológica, o variabilidad entre repeticiones, da como resultado una sobredispersión.
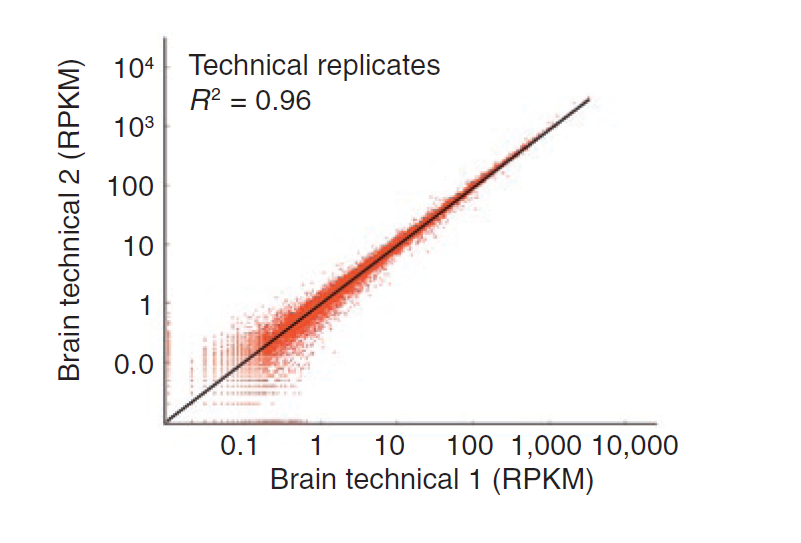
Figura 12.8: La variabilidad técnica sigue una distribución de Poisson
En el caso simple donde tenemos variabilidad entre réplicas, pero sin incertidumbre, podemos mezclar las distribuciones de Poisson de cada réplica en una nueva distribución para modelar la variabilidad biológica. Podemos tratar el parámetro lambda de la distribución de Poisson como una variable aleatoria que sigue una distribución gamma:
\[X \sim \operatorname{Poisson}(\Gamma(r, p)) \nonumber \]
Los recuentos de este modelo siguen una distribución binomial negativa. Para determinar los parámetros para el binomio negativo para cada gen, podemos ajustar una función gamma a través de un diagrama de dispersión de la varianza de conteo promedio vs recuento entre réplicas.
En el caso simple donde hay incertidumbre de mapeo leído, pero no variabilidad biológica, necesitamos incluir la incertidumbre de mapeo en nuestra estimación de varianza. Dado que asignamos lecturas a transcripciones probabilísticamente, necesitamos calcular la varianza en esa asignación.
Los dos hilos de la investigación de análisis de expresión ARN-seq se enfocan en los problemas en estos dos casos simples. Uno de los hilos se enfoca en inferir la abundancia de isoformas individuales para aprender sobre el corte y empalme diferencial y el uso de promotores, mientras que el otro hilo se enfoca en modelar la variabilidad a través de réplicas para crear un análisis de expresión génica diferencial más robusto. Cuffdiff une estos dos hilos separados para estudiar el caso donde tenemos variabilidad biológica y ambigüedad de mapeo de lectura. Dado que la sobredispersión se puede modelar con una distribución binomial negativa y la incertidumbre de mapeo se puede modelar con una distribución Beta, combinamos estas dos para modelar este caso con una distribución binomial beta negativa.