14.5: Reconstrucción
- Page ID
- 54977

La reconstrucción de lecturas es un problema en gran medida estadístico. El objetivo es determinar una puntuación para cada ventana de tamaño fijo en el genoma. Esta puntuación representa la probabilidad de ver el número observado de lecturas dado el tamaño de la ventana. En otras palabras, ¿es poco probable el número de lecturas en una ventana particular dado el genoma? El número esperado de lecturas por ventana se deriva de una distribución uniforme basada en el número total de lecturas (Figura 3). Esta partitura es modelada por una distribución de Poisson.
Sin embargo, este puntaje debe dar cuenta del problema de múltiples hipótesis de prueba, debido a los aproximadamente 150 millones de bases esperadas. Una opción para hacer frente a esto es la corrección Bonferroni, donde el nominal
Figura 14.3: Recuadro 1: ¿Cómo calculamos los QM?


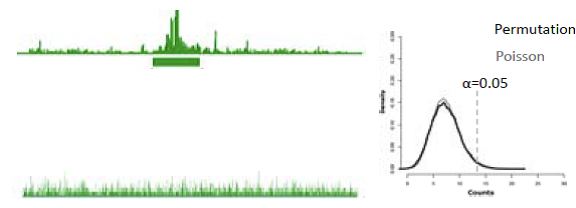
Figura 14.4: Figura 3: La reconstrucción funciona determinando, para una ventana particular, la probabilidad de observar ese número de lecturas (arriba a la izquierda) dada la distribución uniforme del total de lecturas (abajo a la izquierda). Esta probabilidad sigue la distribución de Poisson.
valor p = n * valor p. Este método conduce a una baja sensibilidad, debido a su naturaleza muy conservadora. Otra opción es permutar las lecturas observadas en el genoma, y encontrar el número máximo de lecturas observadas en una sola base. Esto permite un modelo de distribución de conteo máximo, pero el proceso es muy lento. La distribución de escaneo acelera este proceso al computar una forma cerrada para la distribución de conteo máximo para dar cuenta de la dependencia de ventanas superpuestas (Figura 4). La probabilidad de observar k lecturas en una ventana de tamaño w en un genoma de tamaño L dado un total de N lecturas puede aproximarse por [el portaobjetos no está claro].
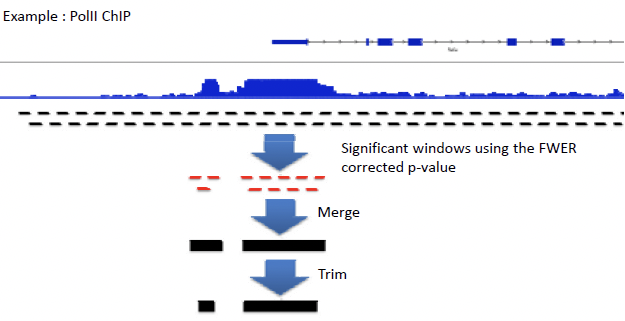
Figura 14.5: Figura 4: Proceso de reconstrucción del genoma a partir de lecturas, utilizando la distribución de exploración
Elegir un tamaño de ventana también es una decisión importante, ya que los genes existen en diferentes niveles de expresión y abarcan diferentes órdenes de magnitud. Las ventanas pequeñas son mejores para detectar regiones puntuadas, mientras que las ventanas más grandes pueden detectar intervalos más largos de mejora moderada. En la mayoría de los casos, se utilizan ventanas de diferentes tamaños para captar señales de tamaño variable.
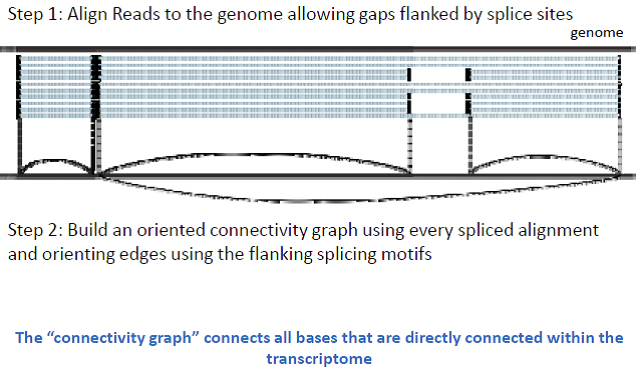
La reconstrucción de la transcripción puede verse como un problema de segmentación, con varios desafíos. Como se mencionó anteriormente, los genes se expresan en diferentes niveles, en varios órdenes de magnitud. Además, las lecturas utilizadas para la reconstrucción se obtienen de ARNm tanto maduro como inmaduro, este último aún conteniendo intrones. Finalmente, muchos genes tienen múltiples isoformas, y la naturaleza corta de las lecturas dificulta la diferenciación entre estos diferentes transcritos. Una herramienta computacional llamada Escritura utiliza el conocimiento a priori de conectividad de fragmentos para detectar transcripciones.
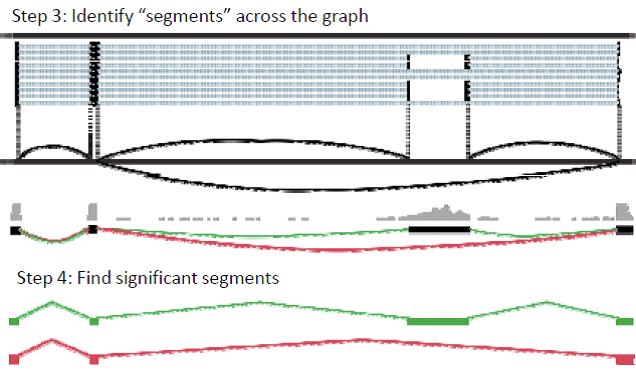
Las isoformas alternativas solo se pueden detectar a través de lecturas de unión de exones, que contienen los extremos de un exón. Las lecturas más largas tienen una mayor probabilidad de abarcar estos cruces (Figura 5). La Escritura trabaja modelando las lecturas usando la estructura gráfica, donde las bases están conectadas a bases vecinas, así como empalmar vecinos. Este proceso difiere de la técnica del gráfico de cadenas, porque se enfoca en el genoma completo, y no mapea secuencias superpuestas directamente. Al deslizar la ventana, las Escrituras pueden saltar a través de uniones de empalme pero aún así examinar isoformas alternativas. A partir de esta gráfica de conectividad orientada, el programa identifica segmentos a través de la gráfica y busca segmentos significativos (Recuadro 2).
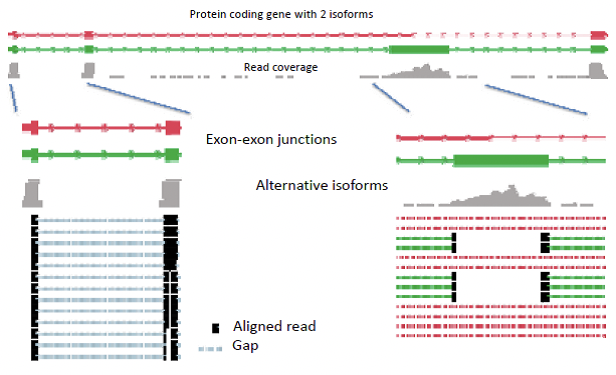
Figura 14.6: Figura 5: Las isoformas alternativas presentan un desafío para la reconstrucción, que debe depender de lecturas que abarcan la unión del exón
El ensamblaje de transcripciones directas es otro método de reconstrucción (a diferencia de los métodos guiados por el genoma como la Escritura). Los métodos de ensamblaje de transcritos son capaces de reconstruir transcritos a partir de organismos sin una secuencia de referencia, mientras que los enfoques guiados por genomas son ideales para anotar genomas de alta calidad y expandir el catálogo de transcritos expresados. Los enfoques híbridos se utilizan para transcritos o transcriptomas de menor calidad que han sido sometidos a reordenamientos importantes, como los de las células cancerosas. Las herramientas populares de ensamblaje de transcripciones incluyen Oasis, Trans-Abyss y Trinity. Otro software popular guiado por genoma es Cufflinks. Independientemente de la metodología o el tipo de software, cualquier experimento de secuenciación que produzca más cobertura genómica experimentará una mejor reconstrucción del transcrito.
Figura 14.7: Recuadro 2: El método de las Escrituras