
15.2: Métodos para medir la expresión génica
- Page ID
- 54994

La forma más intuitiva de investigar un determinado fenotipo es medir los niveles de expresión de proteínas funcionales presentes en un momento dado en la célula. Sin embargo, medir la concentración de proteínas puede ser difícil, debido a sus diferentes ubicaciones, modificaciones y contextos en los que se encuentran, así como por la incompletitud del proteoma. Sin embargo, los niveles de expresión de ARNm son más fáciles de medir y a menudo son una buena aproximación. Al medir el ARNm, analizamos la regulación a nivel de transcripción, sin las complicaciones agregadas de la regulación traduccional y la degradación activa de proteínas, lo que simplifica el análisis a costa de perder información. En este capítulo, consideraremos dos técnicas para generar datos de expresión génica: microarrays y RNA-seq.
Microarrays
Las micromatrices permiten el análisis de los niveles de expresión de miles de genes preseleccionados en un experimento. El principio básico detrás de las micromatrices es la hibridación de fragmentos de ADN complementarios. Para comenzar, segmentos cortos de ADN, conocidos como sondas, se unen a una superficie sólida, comúnmente conocida como chip génico. Luego, la población de ARN de interés, que ha sido tomada de una célula, se transcribe de forma inversa a ADNc (ADN complementario) vía transcriptasa inversa, que sintetiza ADN a partir de ARN usando la cola poli-A como cebador. Para secuencias intergénicas que no tienen cola poli-A, se puede ligar un cebador estándar a los extremos del ARNm. El ADN resultante tiene más complementariedad con el ADN en el portaobjetos que el ARN. El ADNc se lava luego sobre el chip y la hibridación resultante desencadena la fluorescencia de las sondas. Esto se puede detectar para determinar la abundancia relativa del ARNm en la diana, como se ilustra en la figura 15.2.


Figura 15.2: Los valores de expresión génica de experimentos de micromatrices se pueden representar como mapas de calor para visualizar el resultado del análisis de datos.
Actualmente se utilizan dos tipos básicos de microarrays. Los chips génicos de Affymetrix tienen una mancha por cada gen y tienen sondas más largas del orden de 100 nucleótidos. Por otro lado, los conjuntos de oligonucleótidos manchados tejan genes y tienen sondas más cortas alrededor de las decenas de bases.
Existen numerosas fuentes de error en los métodos actuales y los métodos futuros buscan eliminar pasos en el proceso. Por ejemplo, la transcriptasa inversa puede introducir desapareamientos, que debilitan la interacción con la sonda correcta o causan hibridación cruzada, o unión a múltiples sondas. Una solución a esto ha sido usar múltiples sondas por gen, ya que la hibridación cruzada será diferente para cada gen. Aún así, la transcripción inversa es necesaria debido a la estructura secundaria del ARN. La estabilidad estructural del ADN hace que sea menos probable que se doble y no se hibride con la sonda. La siguiente generación de tecnologías, como RNA-seq, secuencian el ARN a medida que sale de la célula, sondeando esencialmente cada base del genoma.
RNA-seq
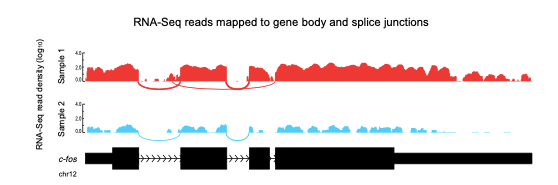
Figura 15.3: lecturas de ARN-seq mapeando a un gen (c-fos) y sus uniones de corte y empalme. Las densidades a lo largo del exón representan densidades de lectura mapeadas a exones (en log10), los arcos corresponden a lecturas de unión, donde el ancho del arco se dibuja en proporción al número de lecturas en esa unión. El gen está regulado a la baja en la Muestra 2 en comparación con la Muestra 1.
RNA-seq, también conocida como secuenciación de escopeta de transcriptoma completa, intenta realizar la misma función que las micromatrices de ADN se han utilizado en el pasado, pero con mayor resolución. En particular, las micromatrices de ADN utilizan sondas específicas, y la creación de estas sondas depende necesariamente del conocimiento previo del genoma y del tamaño de la matriz que se está produciendo. ARN-seq elimina estas limitaciones simplemente secuenciando todo el ADNc producido en experimentos de micromatrices. Esto es posible gracias a la tecnología de secuenciación de próxima generación. La técnica ha sido rápidamente adoptada en estudios de enfermedades como el cáncer [4]. Los datos de RNA-seq se analizan luego agrupando de la misma manera que normalmente se analizarían los datos de las micromatrices.
Matrices de Expresión Génica
Las micromatrices y ARN-seq se utilizan frecuentemente para comparar los perfiles de expresión génica de las células en diversas condiciones. La cantidad de datos generados a partir de estos experimentos es enorme. Las micromatrices pueden analizar miles de genes, y ARN-seq puede, en principio, analizar cada gen que se expresa activamente. El nivel de expresión de cada uno de esos genes se mide a través de una variedad de condiciones, incluyendo cursos de tiempo, etapas de desarrollo, fenotipos, sano vs. enfermo y otros factores.
Para entender lo que transmite el mapa de calor de una matriz de expresión génica (Figura 15.4), primero tenemos que entender lo que nos dice la matriz de datos de expresión. Mediante el uso de microarrays y RNA-seq, podemos obtener el nivel de expresión génica en forma cuantitativa en un experimento. Si tenemos múltiples experimentos, podemos construir una matriz de valores (Figura 15.5) que representa un valor logarítmico de (T/R), donde T es el nivel de expresión génica en la muestra de prueba y R es el nivel de expresión génica en la muestra de referencia.
La matriz de expresión eliminada debido a restricciones de derechos de autor.
Figura 15.4: Transformación de la Figura 4 en un mapa de calor
Si visualizamos la matriz como un mapa de calor, entonces obtenemos la siguiente nueva matriz coloreada:
Estas matrices pueden agruparse jerárquicamente mostrando la relación entre pares de genes, pares de pares, etc., creando un dendrograma en el que se pueden ordenar las filas y columnas usando algoritmos óptimos de ordenación de hojas.
Imagen en el dominio público. Esta gráfica se generó utilizando el programa Cluster from Michael Eisen, que está disponible en Rana.lbl.gov/EisenSoftware.htm, con datos extraídos de la base de datos Stembase de datos de expresión génica.
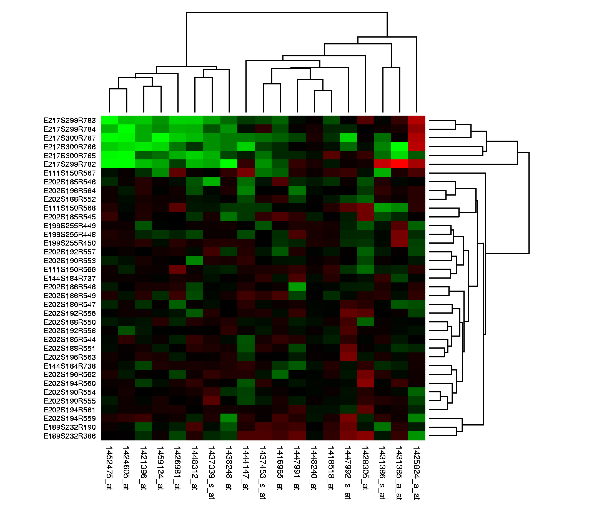
Al revelar la estructura oculta de un largo segmento del genoma, obtenemos una gran comprensión de lo que hace un fragmento de gen, y posteriormente entendemos más sobre la causa raíz de una enfermedad desconocida.
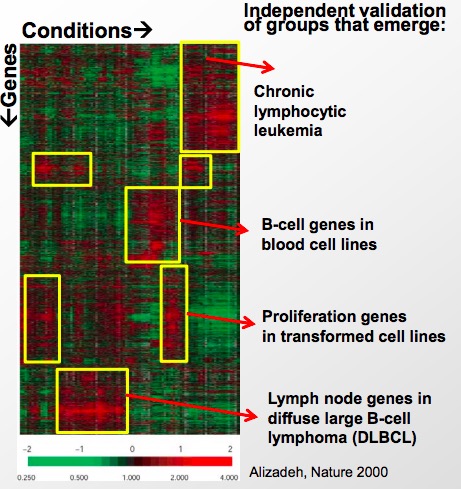
Fuente: Alizadeh, Ash A., Michael B. Eisen, et al. “Distintos tipos de linfoma difuso de células B grandes identificados por perfiles de expresión génica”. Naturaleza 403, núm. 6769 (2000): 503-11.
Figura 15.7: Uso de matriz de expresión génica para inferir más sobre una enfermedad y un segmento génico
Este poder predictivo y analítico se incrementa debido a la capacidad de biclustering de los datos; es decir, agrupamiento a lo largo de ambas dimensiones de la matriz. La matriz permite comparar perfiles de expresión de genes, así como comparar la similitud de diferentes padecimientos como enfermedades. Un reto, sin embargo, es la maldición de la dimensionalidad. A medida que aumenta el espacio de los datos, disminuye el agrupamiento de los puntos. A veces, los datos se pueden reducir a espacios dimensionales más bajos para encontrar estructura en los datos usando agrupamiento para inferir qué puntos pertenecen juntos en función de la proximidad.
Interpretar los datos también puede ser un reto, ya que puede haber otros fenómenos biológicos en juego. Por ejemplo, los exones codificantes de proteínas tienen mayor intensidad, debido a que los intrones se degradan rápidamente. Al mismo tiempo, no todos los intrones son basura y puede haber ambigüedades en el empalme alternativo. También hay mecanismos celulares que degradan los transcritos aberrantes a través de la decadencia mediada sin sentido.