22.5: Métodos Computacionales para Estudiar la Organización del Genoma Nuclear
- Page ID
- 54931

Fuentes de sesgo
Asociación Americana para el Avance de la Ciencia. Todos los derechos reservados. Este contenido está excluido de nuestra
Licencia Creative Commons. Para obtener más información, consulte http://ocw.mit.edu/help/faq-fair-use/.
Fuente: Lieberman-Aiden, Erez, et al. “El mapeo integral de interacciones de largo alcance revela el plegado
Principios del Genoma Humano”. Ciencia 326, núm. 5950 (2009): 289-93.
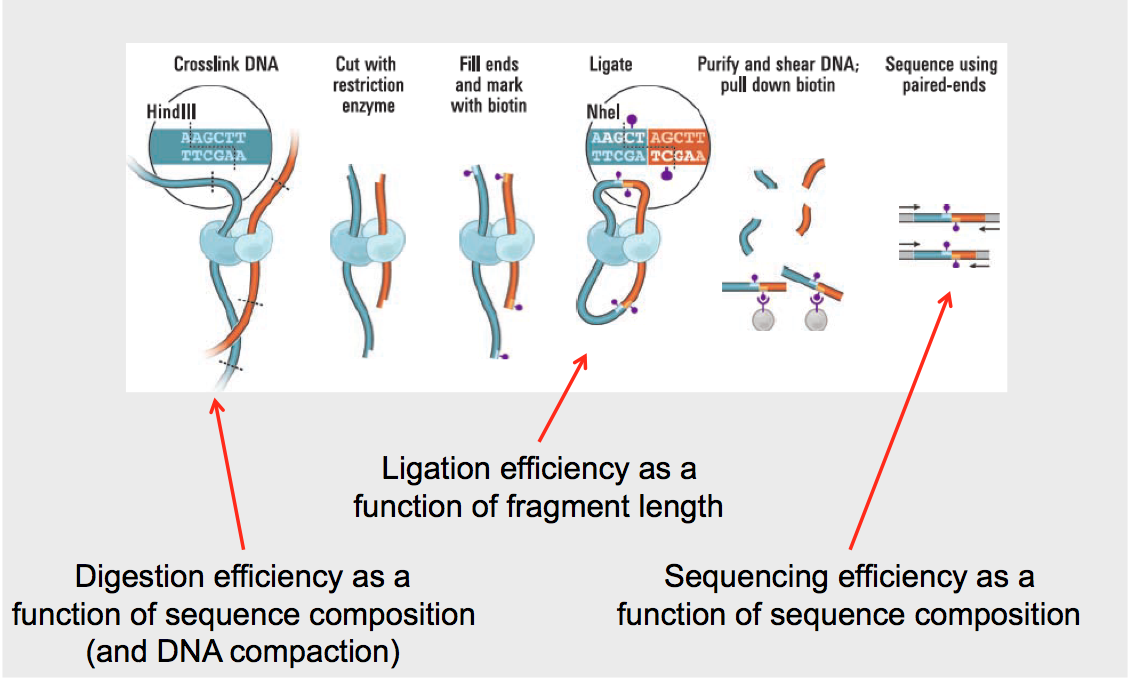
Los tres pasos que potencialmente podrían introducir sesgos incluyen: Digestión, Ligación y Secuenciación. La eficiencia de la digestión es una función de las enzimas de restricción utilizadas y, por lo tanto, algunas regiones del genoma podrían ser menos propensas a ser digeridas ya que su distribución del sitio de reconocimiento particular podría ser realmente escasa. Además, algunas regiones podrían enriquecerse en el sitio de reconocimiento y con ello quedarán sobrerrepresentadas en los resultados. Una solución para esto es usar muchas enzimas de restricción diferentes y comparar los resultados. La eficiencia de ligadura es una función de las longitudes de los fragmentos. Dependiendo de cómo las enzimas de restricción corten la secuencia, algunos extremos pueden tener más o menos probabilidades de ligarse entre sí. Finalmente, la eficiencia de secuenciación es una función de la composición de la secuencia. Algunas cadenas de ADN serán más difíciles de secuenciar, basadas en la riqueza de GC y la presencia de repeticiones, lo que introducirá sesgo.
Corrección de sesgo
Para minimizar el sesgo de ligadura, se eliminan los productos de ligadura no específicos. Dado que los productos de ligación no específicos típicamente tienen sitios de restricción lejanos, introducen fragmentos mucho más grandes. Además, la influencia del tamaño del fragmento en la eficiencia de ligación (Fl en (a len, b len)), la influencia del contenido de G/C en la amplificación y secuenciación (F gc (ag c, b gc)), y la influencia de la singularidad de la secuencia en la mapabilidad (M (a) * M (b)) todos pueden ser contabilizados y corregidos con la ecuación:
P (X a, b) =P anterior * F len (a len, b len) *F gc (a gc, b gc) *M (a) *M (b) Alternativamente, las fuentes de sesgo pueden ser menos explícitamente representadas por la siguiente ecuación:
O i, jj = B i * B j * T i, j
donde la suma de todas las probabilidades de contacto relativas T i, j para cada bin es igual a 1. Los sesgos sólo se asumen como multiplicativos. Esto se resuelve mediante equilibrio de matriz, o ajuste proporcional mediante un algoritmo de corrección iterativa.
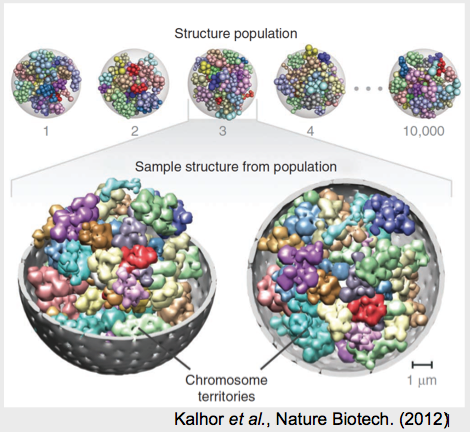
Modelado 3D de datos basados en 3C
El modelado 3D puede revelar muchos principios generales de la organización del genoma. Los modelos actuales se generan usando una combinación de interacciones inter-locus y distancias espaciales conocidas entre hitos nucleares. Sin embargo, queda mucha incertidumbre en los modelos 3D actuales debido a que los datos se recogen de millones de celdas. Los problemas prácticos que afectan al modelado 3D se deben a la gran cantidad de datos necesarios para construir modelos y a las diferentes dinámicas entre una célula individual y una población, que conducen a modelos inestables. El modelado de próxima generación tiende hacia el uso de genómica unicelular.