
27.2: SPIDR
- Page ID
- 54452
Antecedentes
Como se presenta en la información complementaria para SPIDIR, una familia de genes es el conjunto de genes que son descendientes de un solo gen en el ancestro común más reciente (MRCA) de todas las especies bajo consideración. Además, las secuencias genéticas experimentan evolución a múltiples escalas, es decir, a nivel de pares de bases, y a nivel de genes. En el contexto de esta conferencia, dos genes son ortólogos si su MRCA es un evento de especiación; dos genes son parálogos si su MRCA es un evento de duplicación.
En la era genómica, a menudo se conoce la especie de genes modernos; los genes ancestrales se pueden inferir reconciliando árboles de genes y especies. Una reconciliación mapea cada nodo de árbol génico a un nodo de árbol de especies. Una técnica común es realizar la Reconciliación Máxima de Parsimonia (MPR), la cual encuentra la reconciliación R implicando el menor número de duplicaciones o pérdidas utilizando la recursión sobre los nodos internos v de un árbol génico G. MPR puño mapea cada hoja del árbol genético a la hoja de especie correspondiente del árbol de especies. Luego los nodos internos de G se mapean recursivamente:
\[R(v)=\operatorname{MRCA}(R(\operatorname{right}(v)), R(\operatorname{left}(v)))\nonumber\]
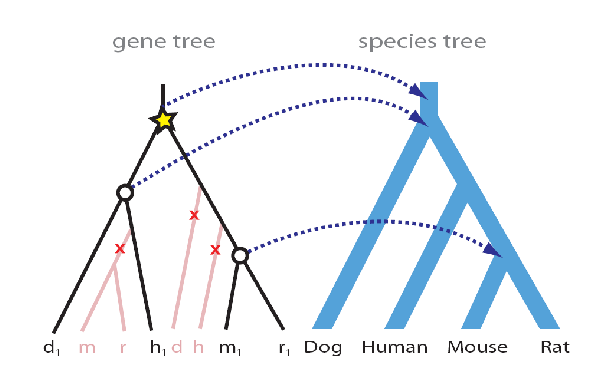
Si un evento de especiación y su nodo ancestral se mapean al mismo nodo en el árbol de la especie. Entonces el nodo ancestral debe ser un evento de duplicación.Usando MPR, la precisión del árbol genético es crucial. Los árboles genéticos subóptimos pueden conducir a un exceso de eventos de pérdida y duplicación. Por ejemplo, si solo una rama está fuera de lugar (como en??) luego la conciliación infiere 3 pérdidas y 1 evento de duplicación. En [6], los autores muestran que los métodos actuales contemporáneos del árbol génico funcionan mal (60% de precisión) en genes individuales. Pero si tenemos genes concatenados más largos, entonces la precisión puede subir hacia el 100%. Además, los genes que evolucionan muy rápida o lentamente portan menos información en comparación con secuencias moderadamente divergentes (40-50% de identidad de secuencia), y tienen un desempeño correspondientemente peor. Como lo corroboran las simulaciones, los genes individuales carecen de información sucia para reproducir la especie correcta de árbol. Los genes promedio son demasiado cortos y contienen muy pocos caracteres filogenéticamente informativos. Mientras que muchos algoritmos de construcción temprana de árboles genéticos ignoraron la información de especies, algoritmos como SPIDIR capitalizan la idea de que el árbol de especies puede proporcionar información adicional que puede ser aprovechada para la construcción de árboles génicos. La sintonía se puede usar para probar independientemente la precisión relativa de las reconstrucciones de árboles génicos dierentes. Esto se debe a que los bloques sinténicos son regiones del genoma donde los organismos recientemente divergentes tienen el mismo orden de genes, y contienen mucha más información que los genes individuales.
Ha habido una serie de algoritmos filogenómicos recientes que incluyen: RIO [2], que utiliza la unión de vecinos (NJ) y el bootstrapping para lidiar con las incogruencias, Orthostrapper [7], que usa NJ y se reconcilia con un árbol de especies vagas, TreeFam [3], que utiliza la curación humana de árboles génicos, así como muchos otros. Varios algoritmos toman una pista más similar a SPIDIR [6], incluyendo [4], un algoritmo de reconciliación probabilística [8], un método bayesiano con reloj, [9], y método de parsimonia usando especies arbóreas, así como desarrollos más recientes: [1] un método bayesiano con reloj relajado y [5], un método bayesiano con tasas relajadas específicas de genes y especies (una extensión a SPIDIR).
Método y Modelo
SPIDIR ejemplifica un algoritmo iterativo para la construcción de árboles génicos usando el árbol de especies. En SPIDIR, los autores definen un modelo generativo para la evolución del árbol genético. Esto consiste en un previo para topología de árbol génico y longitudes de ramas. SPIDIR utiliza un proceso de nacimiento y muerte para modelar duplicaciones y pérdidas (lo que informa al anterior sobre la topología) y luego aprende las tasas de sustitución específicas de genes y especies específicas (que informan la anterior sobre la longitud de las ramas). SPIDIR es un método Maximum a posteriori (MAP) y, como tal, disfruta de varios criterios de optimismo agradables.
En cuanto al problema de estimación, el modelo SPIDIR completo aparece de la siguiente manera:
\[\operatorname{argmax} L, T, R P(L, T, R \mid D, S, \Theta)=\operatorname{argmax} L, T, R P(D \mid T, L) P(L \mid T, R, S, \Theta) P(T, R \mid S, \Theta)\nonumber\]
Los parámetros en la ecuación anterior son: D = datos de alineación, L = longitud de rama T = topología de árbol génico, R = reconciliación, S = árbol de especies (expresado en tiempos),\(\Theta\) = (parámetros específicos de genes y especies [estimados mediante entrenamiento EM],, μ dup/parámetros de pérdida)). Este modelo se puede entender a través de los tres términos en la expresión de la mano derecha, a saber:
- el modelo de secuencia— P (D|T, L). Los autores utilizaron el modelo HKY común para las sustituciones de secuencias, que unifica el modelo de dos parámetros de Kimura para transiciones y transversiones con el modelo de Felsenstein donde la tasa de sustitución depende de la frecuencia de equilibrio de nucleótidos.
- el primer término previo, para el modelo de tasas— P (L|T, R, S,\(\Theta\)), que los autores calculan numéricamente después de aprender las tasas específicas de especies y genes.
- el segundo término previo, para el modelo de duplicación/pérdida— P (T, R|S,\(\Theta\)), que los autores describen mediante un proceso de nacimiento y muerte.
Tener un modelo de tasas es muy útil el modelo de tasas, ya que las tasas de mutación son bastante variables entre los genes. En la conferencia, vimos cómo las tasas estaban bien descritas por una descomposición en tasas específicas de genes y especies. En la conferencia vimos que una distribución gamma inversa parece parametrizar las tasas de sustitución específica del gen, y se nos dijo que una distribución gamma aparentemente captura las tasas de sustitución específica de especies. Contabilizar las tasas específicas de genes y especies permite a SPIDIR construir árboles genéticos con mayor precisión que los métodos anteriores. Se puede elegir un conjunto de entrenamiento para parámetros de tasa de aprendizaje de árboles genéticos que son congruentes con el árbol de la especie. Una preocupación algorítmica importante para las reconstrucciones de árboles genéticos es idear un método de búsqueda rápida de árboles. En la conferencia, vimos cómo la búsqueda en árboles podría acelerarse calculando solo el ArgMaxL completo, T, RP (L, T, R|D, S,\(\Theta\)) para árboles con altas probabilidades previas. Esto se logra a través de un pipeline computacional donde en cada iteración 100s de árboles son propuestos por alguna heurística. La topología anterior P (T, R|D, S,\(\Theta\)) se puede calcular rápidamente. Esto se utiliza como un filtro donde solo se seleccionan las topologías con altas probabilidades previas como candidatas para el cálculo de verosimilitud completa.
El desempeño de SPIDIR se probó en un conjunto de datos reales de 21 hongos. SPIDER recuperó más de 96% de los ortólogos de síntesis mientras que otros algoritmos encontraron menos de 65%. En consecuencia, SPIDER invocó mucho menos número de duplicaciones y pérdidas.