
8.3: Electroforesis
- Page ID
- 53304

La electroforesis utiliza un campo eléctrico aplicado a través de una matriz de gel para separar moléculas grandes como ADN, ARN y proteínas por carga y tamaño. Las muestras se cargan en los pocillos de una matriz de gel que puede separar moléculas por tamaño y se aplica un campo eléctrico a través del gel. Este campo hace que las moléculas cargadas negativamente se muevan hacia el electrodo positivo. La matriz de gel, en sí misma, actúa como un tamiz, a través del cual las moléculas más pequeñas pasan rápidamente, mientras que las moléculas más largas se mueven más lento.
Para el ADN y ARN, clasificar las moléculas por tamaño de esta manera es trivial, debido a la carga negativa uniforme en la cadena principal del fosfato. Para las proteínas, que varían en sus cargas, se debe emplear un truco inteligente para hacerlas imitar a los ácidos nucleicos - ver electroforesis en gel de poliacrilamida (PAGE) a continuación. Diferentes tipos de geles tienen diferentes tamaños de poro. Al igual que los tamices con mallas más finas o más gruesas, algunos geles hacen un mejor trabajo al separar moléculas más pequeñas mientras que otros funcionan mejor para las más grandes. La electroforesis en gel puede usarse como técnica preparativa (es decir, al purificar proteínas o ácidos nucleicos), pero la mayoría de las veces se usa como herramienta analítica.
Electroforesis en gel de agarosa
La electroforesis en gel de agarosa es una técnica utilizada para separar los ácidos nucleicos principalmente por tamaño. La agarosa es un polisacárido obtenido de algas marinas (Figura 8.11). Se puede disolver en tampón de ebullición y verter en una bandeja, donde se instala a medida que se enfría (Figura 8.12) para formar una losa. Los geles de agarosa se vierten con un peine en su lugar para hacer pocillos en los que se colocan muestras de ADN o ARN después de que el gel se haya solidificado. El gel se sumerge en un tampón y se aplica una corriente a través de la losa. El ADN bicatenario tiene una carga negativa uniforme que es independiente de la composición de la secuencia de la molécula. Por lo tanto, si los fragmentos de ADN se colocan en un campo eléctrico migrarán desde el cátodo (-) hacia el ánodo (+). La tasa de migración depende directamente de la capacidad de cada molécula de ADN para gusano o moverse a través del gel de tamizado. La matriz de agarosa proporciona aberturas para que las macromoléculas se muevan. Las macromoléculas más grandes tienen el momento más difícil de navegar a través del gel, mientras que las macromoléculas más pequeñas se deslizan a través de él más rápido.
Figura 8.11 - Estructura del polisacárido de agarosa. Wikipedia
Debido a que la electroforesis utiliza una corriente eléctrica como fuerza para conducir las moléculas a través de la matriz, las moléculas que se están separando deben cargarse. Dado que la relación tamaño/carga para ADN y ARN es constante para todos los tamaños de estos ácidos nucleicos, las moléculas simplemente se clasifican en función de su tamaño, las más pequeñas se mueven más rápido y el movimiento más grande más lento.
Todos los fragmentos de un tamaño dado migrarán la misma distancia sobre el gel, formando las llamadas “bandas” en el gel. La visualización de los fragmentos de ADN en el gel es posible mediante la adición de un colorante, como bromuro de etidio, que se intercala entre las bases y fluoresce cuando se ve bajo luz ultravioleta (Figura 8.13) Al ejecutar ADN de referencia de tamaños conocidos junto con las muestras, es posible determinar los tamaños de los fragmentos de ADN en la muestra. Es útil señalar que, por convención, los fragmentos de ADN no se describen por sus pesos moleculares (a diferencia de las proteínas), sino por su longitud en pares de bases (pb) o kilobases (kb).

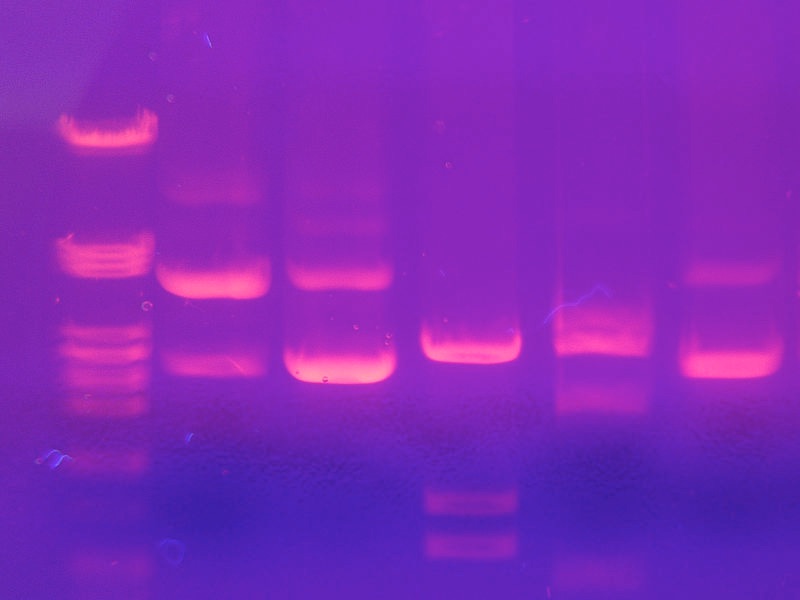
Figura 8.13 - Bandas de ADN visualizadas con tinción con bromuro de etidio. Wikipedia
Electroforesis en gel de poliacrilamida (PAGE)
Al igual que el ADN y el ARN, las proteínas son macromoléculas grandes, pero a diferencia de los ácidos nucleicos, las proteínas no están necesariamente cargadas negativamente. La carga de cada proteína depende de su secuencia de aminoácidos única. Así, las proteínas en una mezcla no necesariamente se moverán todas hacia el ánodo.
Adicionalmente, mientras que el ADN bicatenario tiene forma de varilla, la mayoría de las proteínas son globulares (plegadas). Además, las proteínas son considerablemente más pequeñas que los ácidos nucleicos, por lo que las aberturas de la matriz del gel de agarosa son simplemente demasiado grandes para proporcionar una separación efectiva. En consecuencia, las proteínas inalteradas (nativas) no son muy buenas perspectivas para la electroforesis en geles de agarosa. Para separar las proteínas por masa mediante electroforesis, se deben hacer varias modificaciones.
Matriz de gel
En primer lugar, se emplea una matriz hecha polimerizando y reticulando unidades de acrilamida. Se polimeriza una acrilamida monomérica (Figura 8.14) y los polímeros se reticulan usando N, N'-metilen-bisacrilamida (Figura 8.15) para crear una estructura similar a una malla. Se puede ajustar fácilmente el tamaño de las aberturas de la matriz/malla cambiando el porcentaje de acrilamida en la reacción. Los porcentajes más altos de acrilamida dan aberturas más pequeñas y son más efectivos para separar moléculas más pequeñas, mientras que los porcentajes más bajos de acrilamida se utilizan al resolver mezclas de moléculas más grandes. (Nota: Los geles de poliacrilamida también se utilizan para separar pequeños fragmentos de ácido nucleico, con algunos geles de acrilamida capaces de separar piezas de ADN que difieren en longitud en solo un nucleótido).
Figura 8.15 - Reactivo de reticulación de N, N'-metilenbisacrilamida - acrilamida. Wikipedia
Alteración de carga por SDS
Una segunda consideración es que las proteínas deben alterarse físicamente para “presentarse” a la matriz como las barras de ADN cargadas negativamente. Esto se logra tratando las proteínas con el detergente aniónico, SDS (dodecilsulfato de sodio). El SDS desnaturaliza las proteínas por lo que asumen una forma de varilla y las moléculas de SDS recubren las proteínas de tal manera que la superficie exterior se carga con cargas negativas, enmascarando las cargas originales en las proteínas y haciendo que la carga sobre las proteínas sea más proporcional a su masa, como la columna vertebral del ADN.
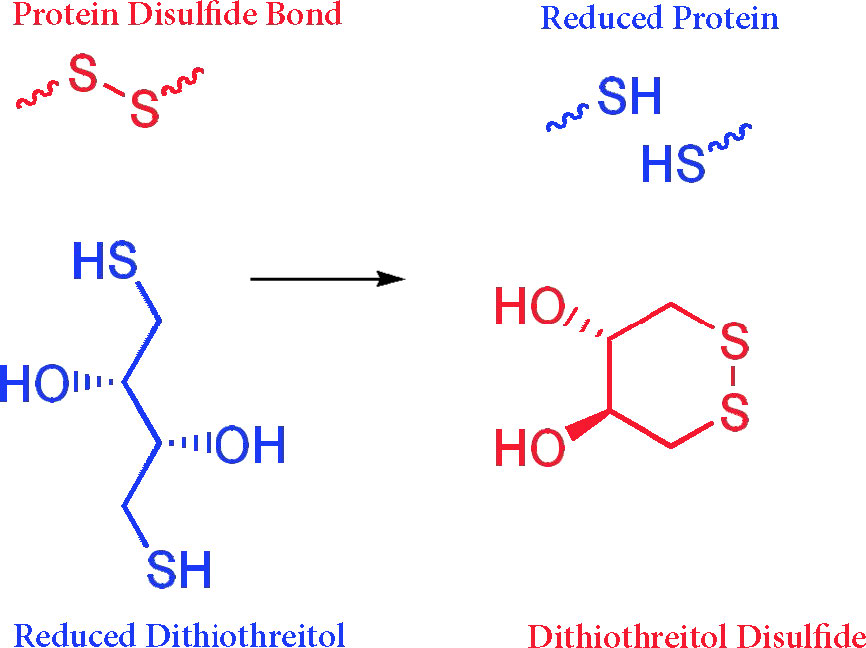
Dado que las proteínas suelen tener enlaces disulfuro que evitan que se desplieguen completamente en el detergente, las muestras se hierven con mercaptoetanol para romper los enlaces disulfuro y garantizar que las proteínas sean lo más parecidas a barras como sea posible en el SDS. Los reactivos como mercaptoetanol (y también ditiotreitol) son reactivos que contienen sulfhidrilo que se oxidan a medida que reducen los enlaces disulfuro en otras moléculas (ver Figura 8.16)
Gel Apilable
Una tercera consideración es que se puede emplear un “gel de apilamiento” en la parte superior de un gel de poliacrilamida para proporcionar una manera de comprimir las muestras en una banda estrecha antes de que entren en el gel de poliacrilamida principal (llamado el gel de resolución). Al igual que los fragmentos de ADN en la electroforesis en gel de agarosa se clasifican en función del tamaño (el movimiento más grande más lento y el más pequeño se mueven más rápido), las proteínas migran a través de la matriz de gel a velocidades inversamente relacionadas con su tamaño. Al finalizar la electroforesis, las proteínas pueden visualizarse mediante tinción con compuestos que se unen a proteínas, como Coomassie Brilliant Blue (Figura 8.17) o nitrato de plata.
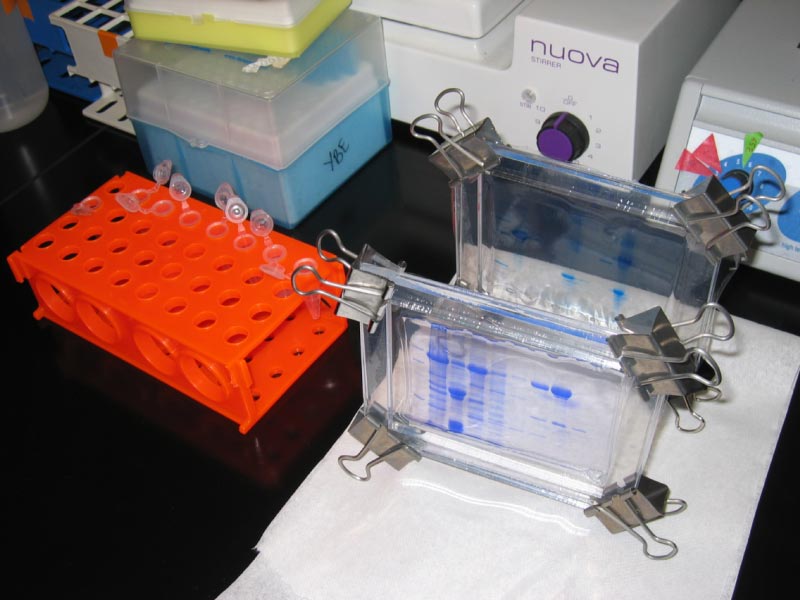
Figura 8.17 - Dos geles SDS-PAGE - Las proteínas son las bandas azules (teñidas con Azul Coomassie). Wikipedia
Electroforesis en gel no desnaturalizante
La técnica SDS_PAGE descrita anteriormente es el método más común utilizado para la separación electroforética de proteínas. En algunas situaciones, sin embargo, las proteínas pueden resolverse en los llamados geles “nativos”, en ausencia de SDS. Bajo estas condiciones, el movimiento de las proteínas a través del gel se verá afectado no simplemente por su masa, sino por su carga al pH del gel, también. Las proteínas complejadas con otras moléculas pueden moverse como una sola entidad, permitiendo el aislamiento de las parejas de unión de proteínas de interés.
Enfoque isoeléctrico
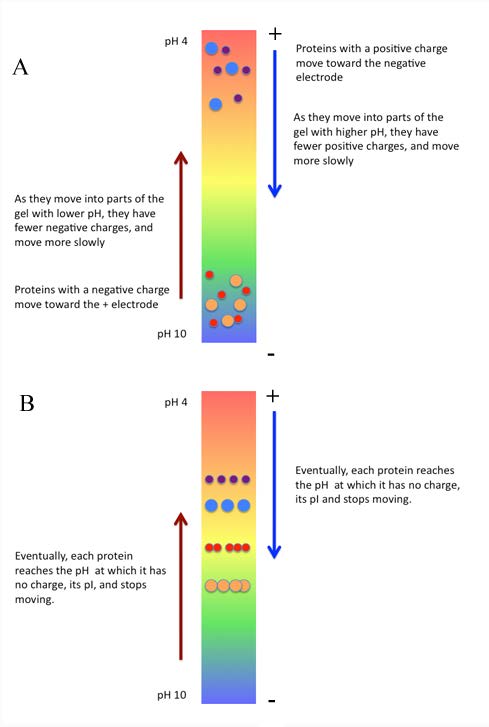
Las proteínas varían considerablemente en sus cargas y, consecuentemente, en sus valores de pI (pH al que su carga es cero). Esto se puede explotar para separar las proteínas en una mezcla. La separación de proteínas mediante enfoque isoeléctrico requiere el establecimiento de un gradiente de pH en un tubo que contiene una matriz de gel de acrilamida. El tamaño de poro del gel se ajusta para que sea grande, para reducir el efecto del tamizado basado en el tamaño. Las moléculas a separar se aplican al gel que contiene el gradiente de pH y se aplica un campo eléctrico. Bajo estas condiciones, las proteínas se moverán de acuerdo a su carga.
Las moléculas cargadas positivamente, por ejemplo, se mueven hacia el electrodo negativo, pero como están viajando por un gradiente de pH, a medida que pasan por él, alcanzan una región donde su carga es cero y, en ese punto, dejan de moverse. En ese punto no se sienten atraídos por el electrodo positivo ni el negativo y, por lo tanto, son “enfocados” en su pI (Figura 8.18). Mediante el enfoque isoeléctrico, es posible separar proteínas cuyos valores de pI difieren en tan solo 0.01 unidades.
Electroforesis en gel 2D
Tanto el SDS-PAGE como el enfoque isoeléctrico son técnicas poderosas, pero una combinación inteligente de las dos es una poderosa herramienta de proteómica, la ciencia de estudiar todas las proteínas de una célula/tejido simultáneamente. En la electroforesis en gel 2-D, primero se prepara un lisado a partir de las células de interés. Las proteínas en el lisado se separan primero por su pI, mediante enfoque isoeléctrico y luego por tamaño por SDS-PAGE.

La mezcla de proteínas se aplica primero a un tubo o tira (Figura 8.19, Etapa 1) donde se realiza isoelectroenfoque para separar las proteínas por sus valores de pI (Paso 2). A continuación, como se muestra en la figura, el gel que contiene las proteínas separadas por sus IP se da vuelta de lado y se aplica a lo largo de la parte superior de una losa de poliacrilamida para que la SDS-PAGE se separe en base al tamaño (Etapa 3). Las proteínas en la matriz de enfoque isoeléctrico se someten a electroforesis en el gel de poliacrilamida y se separan en función del tamaño. El producto de este análisis es un gel 2-D como se muestra en la Figura 8.20.El poder de la electroforesis en gel 2-D es que prácticamente todas las proteínas de una célula pueden separarse y aparecer en el gel como una mancha definida por su tamaño único y pI. En la figura, las manchas en la parte superior izquierda corresponden a proteínas grandes con carga positiva, mientras que las de la parte inferior derecha son pequeñas con carga negativa. Cada mancha en un gel 2-D puede eluirse e identificarse mediante el uso de espectrometría de masas de alto rendimiento. Esto es particularmente poderoso cuando se comparan perfiles de proteínas entre diferentes tejidos o entre muestras control y tratadas del mismo tejido.

Figura 8.20 - Resultado de la separación por electroforesis en gel 2-D. Wikipedia
Comparación de perfiles de proteínas
La comparación de geles 2-D de proteínas de tejido no canceroso y proteínas de un tejido canceroso del mismo tipo proporciona una identificación rápida de proteínas cuyo nivel de expresión difiere entre las dos. Información como esta puede ser útil en el diseño de tratamientos o en la comprensión del mecanismo o mecanismos por los cuales se desarrolla el cáncer.