
3.3: Reacción en cadena de la polimerasa (PCR) y clonación de productos de PCR
- Page ID
- 57259

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
La PCR es una técnica in vitro para la amplificación de una región de ADN que se encuentra entre dos regiones de secuencia conocida. La amplificación por PCR se logra mediante el uso de cebadores de oligonucleótidos. Estos son típicamente oligonucleótidos monocatenarios cortos que son complementarios a las regiones externas de secuencia conocida.
.png)
Figura 3.3.1: Cebadores de igonucleótidos ol complementarios
Los oligonucleótidos sirven como cebadores para la ADN polimerasa y las cadenas desnaturalizadas del fragmento de ADN grande sirven como molde. Esto da como resultado la síntesis de nuevas cadenas de ADN que son complementarias a las cadenas molde parentales. Estas nuevas cadenas tienen extremos 5' definidos (los extremos 5' de los cebadores oligonucleotídicos), mientras que los extremos 3' son potencialmente ambiguos en longitud.
.png)
Figura 3.3.2: Síntesis de nuevas cadenas de ADN
La síntesis dirigida a oligonucleótidos de cadenas de ADN hijas puede repetirse si el nuevo dúplex se desnaturaliza (por calentamiento) y se permite que los cebadores adicionales se hibriden (enfriando a una temperatura apropiada).
Los pasos de:
- Desdesnaturalización de la plantilla
- Recocido de cebadores
- Extensión de imprimación
comprender un único “ciclo” en la metodología de amplificación por PCR.
Después de cada ciclo, las cadenas de ADN recién sintetizadas pueden servir como moldes en el siguiente ciclo.
.png)
Figura 3.3.3: Producto de la reacción de elongación como molde
Nótese que la mitad de las cadenas recién sintetizadas de la segunda ronda de replicación tienen extremos 5' y 3' que están definidos por la ubicación de hibridación de los oligonucleótidos cebadores.
Resumen de productos al final de cada ciclo de PCR:
.png)
Figura 3.3.4: Productos de PCR
El total de cada uno de los diferentes tipos de fragmentos de oligonucleótidos para cada ciclo se puede resumir de la siguiente manera:
.png)
Figura 3.3.5: Tipos de fragmentos para cada ciclo
- La amplificación de los fragmentos sigue el siguiente patrón:
- Siempre hay 1x copia de cada una de las plantillas originales - la PCR como se describe nunca reproduce la plantilla de longitud completa
- Habrá 'n'x copias de cada fragmento con una longitud indeterminada (donde 'n' es el número de ciclos). Los fragmentos de longitud indeterminada tienen un extremo definido por un cebador de PCR y el otro extremo es de longitud indeterminada
- Habrá (2 n - (n + 1)) x copias de cada fragmento de longitud definida (es decir, cada extremo definido por los dos cebadores de PCR).
- Por ejemplo, después de 6 ciclos tendremos
- 1 x copia de cada plantilla original
- 6 x copias de cada fragmento de longitud indeterminada
- (64 - (6 + 1)) = 57 x copias de cada fragmento de longitud definida
- En el caso anterior, el producto de PCR deseado será un dúplex del fragmento de longitud definida. La pregunta es: ¿cuántos se producirán?
- Las plantillas originales no necesariamente se recocerán entre sí
- Del mismo modo, las plantillas de longitud indeterminada no necesariamente se recocerán entre sí
- Además, a medida que avanza el número de ciclos, los fragmentos de longitud definida superan con creces a los demás
- Por lo tanto, lo más probable es que el molde original y los fragmentos de longitud indeterminada se hibriden con fragmentos de longitud definida.
- Nuestra amplificación esperada sería, por lo tanto, (usando el ejemplo de PCR de 6 ciclos)
57 - (6 + 1) = 50 x copias de cada fragmento de longitud definida
(es decir, 50 x dúplexes de longitud definida)
- La amplificación esperada del producto de longitud definida deseada con respecto a la concentración de molde original (\(x\)) puede representarse así por la fórmula:
\[[(2^n - (n + 1)) - (n + 1)] x\]
o
\[(2^n - 2(n + 1))\]
(esto a menudo se abrevia a una simple regla general para la amplificación: (2 n - 2n) x)
La interpretación de esta fórmula es que
- Para un número dado de ciclos 'n' hacemos '2 n x' totales posibles dúplex
- Para un número dado de ciclos habrá '2 (n+1) (o 2n en nuestra aproximación) x' dúplexes que se forman a partir de la plantilla original, o un fragmento de longitud indeterminada, junto con un fragmento de longitud definida (y representan un producto no deseado)
- Así, la concentración total del producto deseado (dúplex con una longitud definida por los cebadores de PCR) será
(2 n - 2 (n+1)) x (donde x es la concentración del dúplex original) - El valor teórico de amplificación nunca se alcanza en la práctica. Varios factores impiden que esto ocurra, entre ellos:
- La competencia de cadenas hijas complementarias con cebadores para la rehibridación (es decir, la rehibridación de dos cadenas hijas no produce amplificación).
- Pérdida de actividad enzimática por desnaturalización térmica, especialmente en los ciclos posteriores
- Incluso sin desnaturalización térmica, la cantidad de enzima se vuelve limitante debido al exceso de diana molar en ciclos posteriores (es decir, después de 25 a 30 ciclos, muchos cebadores necesitan extenderse)
- Posible hibridación del cebador del segundo sitio y cebado no productivo
Nota
La PCR fue inventada en 1985 por Kary Mullis, trabajando para la corporación Cetus en algún lugar cerca de Berkeley, California. El método original de PCR utilizó el fragmento Klenow de la ADN polimerasa I. Esta enzima, sin embargo, se desnaturaliza a temperaturas inferiores a las requeridas para desnaturalizar la mayoría de los dúplex molde. Así, después de cada ciclo, hubo que añadir enzima fresca a la reacción. Esto fue bastante tedioso. Además de este problema con la enzima, las muestras tuvieron que trasladarse de un baño de temperatura a otro para permitir las etapas individuales de desnaturalización, hibridación y polimerización, las cuales requirieron diferentes temperaturas. Esto también fue bastante tedioso.
Así, dos avances principales permitieron automatizar el proceso, estos avances fueron:
- El uso de ADN polimerasas termoestables, que resistieron la desnaturalización (inactivación) a altas temperaturas. Así, una alícuota inicial de polimerasa podría durar a lo largo de los numerosos ciclos del protocolo. La primera ADN polimerasa termoestable que se utilizó fue aislada de la bacteria Thermus aquaticus. Se aisló de una fuente termal en el Parque Nacional Yellowstone donde vivió felizmente (es decir, replicó su ADN) a temperaturas superiores a 85 °C
- El desarrollo de baños de temperatura que podrían cambiar sus temperaturas arriba y abajo rápidamente y de manera automatizada y programada. Estos se conocen como termocicladores o máquinas de PCR.
Estos dos desarrollos permiten la automatización de la PCR. El proceso de PCR está cubierto por patentes propiedad de Hoffmann-La Roche Inc. (un conglomerado sin rostro en el que puedes confiar).
Parámetros de ciclo térmico
- Los parámetros de ciclado térmico son críticos para un experimento de PCR exitoso. Los pasos importantes en cada ciclo de PCR incluyen:
- desnaturalización de plantilla
- hibridación de cebadores
- extensión de los cebadores
Un perfil de temperatura representativo para cada ciclo podría tener el siguiente aspecto:
.png)
Figura 3.3.6: Temperatura para el ciclo de PCR
Desnaturalización de la plantilla
- La desnaturalización inicial del molde se realiza a 95-100 °C.
- Los plásmidos superenrollados son más difíciles de fundir y pueden requerir ebullición durante varios minutos, o pueden desnaturalizarse inicialmente usando base (NaOH, seguido de neutralización del pH).
- La desnaturalización durante el experimento de PCR (es decir, el segundo ciclo en adelante) generalmente se realiza a temperaturas de 92-95 °C (generalmente determinadas empíricamente).
Temperatura de recocido del cebador
- La temperatura de reasociación del cebador es un parámetro importante en el éxito del experimento de PCR.
- La temperatura de hibridación es característica para cada oligonucleótido:
- es una función de la longitud y composición de bases del cebador así como de la fuerza iónica del tampón de reacción.
- Las estimaciones de la temperatura de recocido se pueden calcular de varias maneras diferentes.
- Estas temperaturas de hibridación calculadas son un punto de partida para el experimento de PCR, pero las temperaturas de hibridación ideales se determinan empíricamente.
Extensión de imprimación
- La extensión del cebador generalmente se realiza a 72 °C, o la temperatura óptima de la ADN polimerasa.
- La longitud de tiempo de las etapas de extensión del cebador puede aumentarse si la región del ADN a amplificar es larga, sin embargo, para la mayoría de los experimentos de PCR un tiempo de extensión de 2 minutos es suficiente para obtener la extensión completa.
Número de ciclos
- El número de ciclos suele estar entre 25 y 35.
- Más ciclos significan un mayor rendimiento de producto.
- Sin embargo, al aumentar el número de ciclos, mayor es la probabilidad de generar diversos artefactos (por ejemplo, productos de cebado erróneo).
- Es inusual encontrar procedimientos que tengan más de 40 ciclos.
Elección de polimerasas para PCR
- Uno de los avances importantes que permitieron el desarrollo de la PCR fue la disponibilidad de polimerasas termoestables.
- Esto permitió que la enzima inicialmente añadida sobreviviera a ciclos de temperatura cercanos a los 100 °C.
ADN polimerasas termoestables y sus fuentes
|
ADN Polimerasa |
Natural o recombinante |
Fuente |
|---|---|---|
|
Taq |
Natural |
Thermus aquaticus |
|
Amplitaq |
Recombinante |
T. aquaticus |
|
Amplitaq (fragmento Stoffel)® |
Recombinante |
T. aquaticus |
|
Bañera de hidromasaje™ |
Natural |
Thermus flavis |
|
Pirostasa™ |
Natural |
T. flavis |
|
Ventilación™ |
Recombinante |
Thermococcus litoralis |
|
Ventilación Profunda™ |
Recombinante |
Pyrococcus GB-D |
|
Tth |
Recombinante |
Thermus thermophilus |
|
PFU |
Natural |
Pyrococcus furiosus |
|
UltMA™ |
Recombinante |
Thermotoga maritima |
- Propiedades de las ADN polimerasas utilizadas en PCR
|
|
Taq /Amplitaq |
Fragmento de Stoffel |
Ventilación™ |
Ventilación Profunda™ |
PFU |
Tth |
UltMA™ |
|---|---|---|---|---|---|---|---|
|
Vida media de 95 °C |
40 min |
80 min |
400 min |
1380 min |
>120 min |
20 min |
>50 min |
|
5'3' exo |
+ |
+ |
|||||
|
3'5' exo |
+ |
+ |
+ |
+ |
|||
|
Velocidad de extensión (nt/seg) |
75 |
>50 |
>80 |
? |
60 |
>33 |
? |
|
Actividad de RT |
Débil |
Débil |
? |
? |
? |
Sí |
? |
|
Finales resultantes |
3' A |
3' A |
> 95% contundente |
> 95% contundente |
contundente |
3' A |
contundente |
|
Desplazamiento del hilo |
+ |
+ |
|||||
|
M.W. (kDa) |
94 |
61 |
? |
? |
92 |
94 |
70 |
Bufferes y MgCl 2 en reacciones de PCR
Un tampón de reacción típico para PCR sería algo así como:
- Tris 10 mM, pH 8.3
- 50 mM KCl
- 1.5 mM MgCl 2
- Gelatina al 0.01%
- La concentración de MgCl 2 en la mezcla de reacción final suele estar entre 0.5 y 5.0 mM, y la concentración óptima se determina empíricamente (típicamente entre 1.0 - 1.5 mM). Iones Mg 2+:
- formar un complejo soluble con dNTP que es esencial para la incorporación de dNTP
- estimular la actividad de la polimerasa
- aumentar la Tm (temperatura de fusión) de la interacción cebador/molde (es decir, sirve para estabilizar la interacción dúplex
En general,
- bajo Mg 2+ conduce a rendimientos bajos (o ningún rendimiento) y
- alto Mg 2+ conduce a la acumulación de productos inespecíficos (cebado erróneo).
Impres
Diseño de imprimación
- Generalmente, los cebadores utilizados son de 20 a 30 mer de longitud. Esto proporciona temperaturas prácticas de hibridación (del régimen de alta temperatura donde la polimerasa termoestable es más activa).
- Los cebadores deben evitar tramos de secuencias de polibases (por ejemplo, poli dG) o motivos repetitivos, estos pueden hibridarse con registros inapropiados en el molde.
- Deben evitarse las secuencias repetidas invertidas para evitar la formación de estructura secundaria en el cebador, lo que impediría la hibridación con el molde.
- Deben evitarse las secuencias complementarias a otros cebadores utilizados en la PCR para evitar la hibridación entre cebadores (particularmente importante para el extremo 3' del cebador)
- Si es posible, el extremo 3' del cebador debe ser rico en bases G, C para mejorar la hibridación del extremo que se extenderá.
- La distancia entre los cebadores debe ser inferior a 10 Kb de longitud. Típicamente, se observa una reducción sustancial en el rendimiento cuando los cebadores se extienden entre sí más allá de ~3 Kb.
Temperatura de fusión (Tm) de las imprimaciones
- La Tm de hibridación del cebador se puede calcular usando diversas fórmulas. La fórmula más utilizada es:
\[Tm = [(number of A+T residues) \times 2 °C] + [(number of G+C residues) x 4 °C]\]
- Esta fórmula se determinó originalmente a partir de ensayos de hibridación de oligonucleótidos, los cuales se realizaron en NaCl 1 M, y parece ser precisa en condiciones de menor sal solo para cebadores de menos de aproximadamente 20 nucleótidos de longitud.
- La sabiduría común es que la Tm es más como 3-5 °C menor que el valor calculado a partir de esta fórmula.
\[Tms = 81.5 + 16.6(\log_{10}[J+]) + 0.41(\%G+C) - (600/l)\]
- Donde [J +] = la concentración molar de cationes monovalentes (e.g. Na + de NaCl), y l = la longitud del oligonucleótido. (%G+C) es el valor porcentual real y no una representación fraccional (es decir, el valor a insertar para una imprimación que tenía un contenido de 90% de G+C sería “90" y no “0.90").
- Según se informa, esta fórmula es útil para cebadores de 14 a 70 bases de longitud.
\[Tmp = 22 + 1.46([2 \times (G+C)] + (A+T))\]
- Según se informa, esta fórmula es útil para cebadores de 20-35 bases de longitud.
- La temperatura de recocido calculada es solo una temperatura de referencia a partir de la cual iniciar experimentos.
- La temperatura real de recocido puede ser 3-12 °C mayor que la Tm calculada.
- La condición real de la temperatura de recocido debe determinarse empíricamente.
- Se debe usar la temperatura de recocido más alta que dé el mejor producto de PCR.
- Ejemplos de cálculos de T m, T m s y T m p (0.05 M K +)
|
|
G |
A |
T |
C |
T m |
T m |
T m p |
|---|---|---|---|---|---|---|---|
|
15 mer |
3 |
5 |
2 |
5 |
46 |
42 |
56 |
|
20 mer |
6 |
5 |
4 |
5 |
62 |
52 |
67 |
|
30 mer |
8 |
6 |
8 |
8 |
92 |
62 |
89 |
Cálculo de concentraciones de imprimación
- La concentración molar de un cebador se puede calcular con base en la absorbancia del cebador a 260 nm (A 260) y el coeficiente de extinción molar para el cebador a esta longitud de onda.
- El coeficiente de extinción molar para el cebador se puede calcular conociendo la secuencia del cebador y luego sumando los valores de extinción molar para las bases individuales que comprenden el cebador.
- Las bases individuales tienen los siguientes coeficientes de extinción molar a 260 nm:
- Una solución 1.0 molar de dT tiene un valor de 8.400 unidades de absorbancia a 260 nm.
- Una solución 1.0 molar de dA tiene un valor de 15.200 unidades de absorbancia a 260 nm.
- Una solución 1.0 molar de dG tiene un valor de 12,010 unidades de absorbancia a 260 nm.
- Una solución 1.0 molar de dC tiene un valor de 7050 unidades de absorbancia a 260 nm.
- Por ejemplo, el cebador 5' TAGC 3' tendría un coeficiente de extinción molar de 42,660 a 260 nm. Asimismo, una solución 10 micromolar de este cebador daría una absorbencia de 0.427 a 260 nm.
Clonación de productos PCR
Introducción de sitios de restricción
- Es posible introducir secuencias de sitios de restricción en productos de PCR incorporando estas secuencias en el extremo 5' del cebador o cebadores de PCR.
.png)
Figura 3.3.7: Introducción de la secuencia del sitio de restricción
- La secuencia corta del sitio de restricción en el extremo 5' del cebador de PCR no hibridará, pero siempre que la región de hibridación 3' sea lo suficientemente larga (es decir, su Tm es lo suficientemente alta; ~20 meros) el cebador se unirá específicamente al sitio apropiado.
- El producto de PCR tendrá así una secuencia de ADN adicional en el extremo 5' que contendrá el sitio de restricción de endonucleasa.
- Se puede agregar una secuencia de sitio de restricción similar o diferente a través del otro cebador de PCR.
- Si el otro cebador tiene una secuencia de restricción diferente entonces el fragmento de PCR puede insertarse de una manera dependiente direccional en un plásmido huésped.
Los problemas potenciales con este método incluyen:
- No hay una manera fácil de evitar que se corten sitios internos que contienen secuencias de restricción similares cuando se corta el final del producto de PCR.
- Las secuencias de restricción son secuencias de repetición inversa, por lo que existe el potencial para la asociación del dímero del cebador y la hibridación no productiva resultante
Generación de medios sitios
- Este método es similar al método de introducción de sitios de restricción, descrito anteriormente.
- La principal diferencia es que en lugar del cebador que contiene toda la secuencia del sitio de restricción (digamos los seis nucleótidos de un cortador de seis) contendrá solo los tres últimos (y el otro cebador de PCR contendrá la secuencia complementaria para los tres primeros).
.png)
Figura 3.3.8: Mitad de sitios
Las ventajas de este método son:
- Típicamente, los sitios de restricción internos se escinden con una eficiencia mucho mayor (es decir, algunos sitios si se encuentran en los extremos del ADN lineal nunca cortan bien en absoluto)
- No hay necesidad de purificar en gel los fragmentos enlazadores después de la digestión
- El ADN puede ser metilado (los medios sitios no lo serán). Después de la concatenación se cortarán los enlazadores pero los sitios de restricción internos no
- Una desventaja es que el mismo sitio de restricción se incorpora en ambos extremos de manera que el fragmento de PCR no puede ligarse en un vector huésped de una manera dependiente de la orientación.
- Además, en este método el saliente 3' A no puede ser tolerado.
Ligadura de extremos romos
- Como se indicó anteriormente, algunas ADN polimerasas termoestables añaden un único residuo dA en el extremo 3' del producto de PCR.
- Hay tres elecciones a tomar cuando se intenta subclonar sin el uso de sitios de restricción añadidos dentro de los cebadores:
- Usar una ADN polimerasa que deje la cadena 3' roma (por ejemplo, Vent) y haga una ligadura de extremo romo (es decir, el vector hospedador se abrió con endonucleasa de restricción de corte romo)
- “Arreglar” el saliente 3' A masticando de nuevo con Pol I, dNtP's.
- Use el saliente 3' A para recocer y ligar a un vector “T”, un vector que tiene un único saliente dT en sus extremos 3'.
- “Los vectores “" T "” se pueden hacer abriendo un vector, a través de una endonucleasa de restricción de corte romo y ligando en un enlazador específico.”
- El enlazador contiene una secuencia de restricción para una endonucleasa de restricción que reconoce un palíndromo interrumpido y corta en la región interna con un único saliente 3'.
- El enlazador contendrá dos copias de la secuencia de reconocimiento de restricción, la primera con una secuencia en el palíndromo interrumpido que deja un saliente 3' T y la segunda con una secuencia en el palíndromo interrumpido que deja otro saliente 3' T
- Esto puede sonar confuso, así que aquí hay un ejemplo de cómo hacer un vector T:
Endonucleasa de restricción Ahd I:
|
G A C N N N N N G T C
C T G N N N N N C A G
|
Ahd voy a cortar esta secuencia para producir:
G A C N N N N N G T C C T G N N N N N C A G
Podríamos diseñar un oligonucleótido con dos secuencias de restricción Ahd I, con secuencias ligeramente diferentes en la región interrumpida del palíndromo, para dar:
| |
G A C N N T N N G T C G A C N N A N N G T C
C T G N N A N N C A G C T G N N T N N C A G
| |
Si esto se insertara en un vector, y el vector después se cortara con Ahd I, tendría la siguiente secuencia en los extremos del vector linealizado:
-G A C N N T N N G T C- -C T G N N T N N C A G-
En otras palabras, un saliente 3' 'T' en ambos extremos del vector. Un producto de PCR, con voladizos 3' A podría insertarse así en dicho vector 'T'
Agregar promotores, sitios de unión a ribosomas, codones de inicio y codones de parada
- La capacidad de añadir secuencias únicas a los extremos 5' de los cebadores de PCR permite incorporar directamente elementos de control cortos.
- Estos pueden incluir un codón de inicio o codón de terminación (3 bases), un promotor (región de ~30 nucleótidos) o un sitio de unión al ribosoma (~8 bases).
.png)
Figura 3.3.9: Inserción de promotor/ribosoma/sitio de unión/codón de inicio/codón de terminación
Muagénesis por PCR
Fusión génica
- Este método es útil para unir regiones superpuestas de un gen grande, o para la construcción de genes quiméricos.
.png)
Figura 3.3.10: Fusión génica
Creación de deleciones dentro de un gen
- Se puede usar una metodología muy similar dentro de un solo gen para la producción de un gen mutante que contiene una deleción específica:
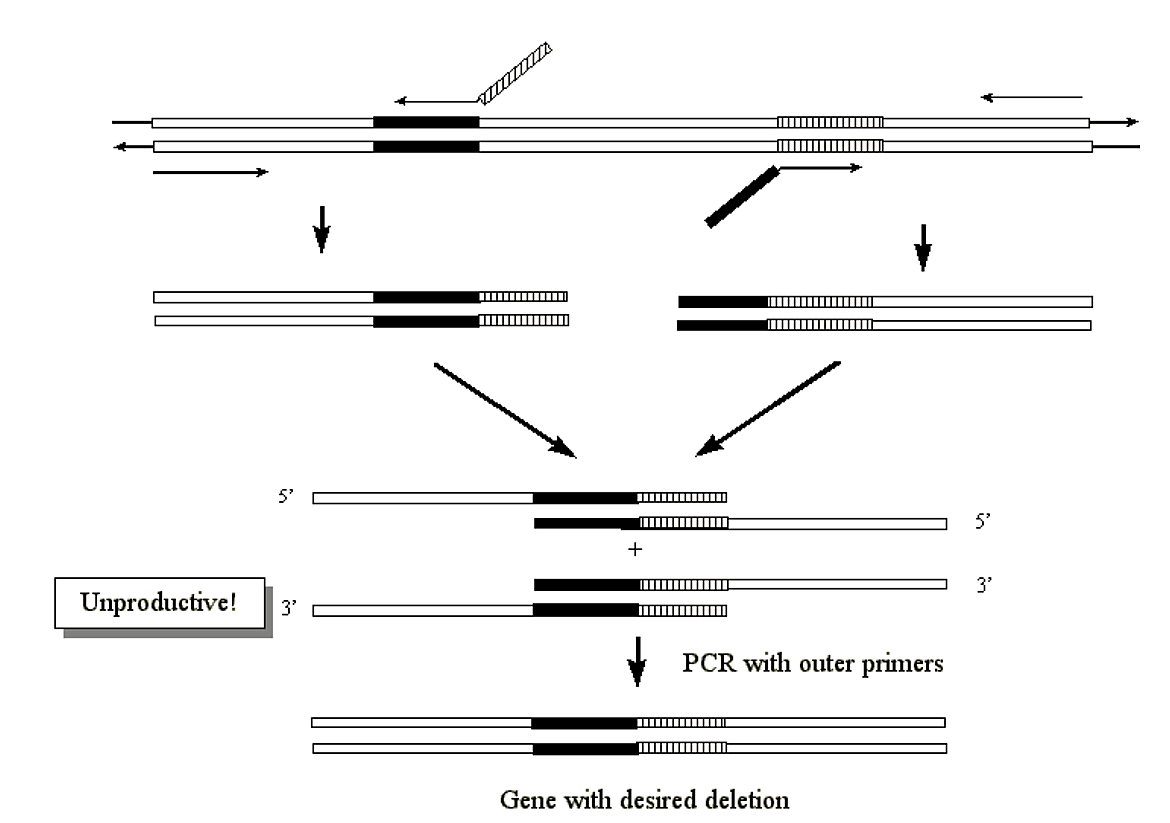.png)
Figura 3.3.11: Supresiones
- Si el gen está contenido dentro del ADN circular (es decir, un plásmido), se pueden construir deleciones en una sola reacción de PCR con un solo conjunto de cebadores (este tipo de metodología también se conoce como PCR “inversa”).
.png)
Figura 3.3.12: PCR inversa
Generación de mutación (es) puntual (es decir, mutaciones de sustitución de bases)
- La generación de sustituciones de bases puede proceder a lo largo de una ruta similar a la de las mutaciones de deleción.
- Sin embargo, en este caso los cebadores son mutagénicos - habrá un desajuste, o desapareamientos, entre las secuencias de cebador y diana.
- El oligo mutagénico tendrá una Tm menor a la esperada debido a este desapareamiento (es).
.png)
Figura 3.3.13: Mutación puntual
Introducción de sustituciones de bases mediante PCR asimétrica:
.png)
Figura 3.3.14: Sustitución con PCR asimétrica
Mutágenesia de inserción
- Las inserciones cortas (~1-6 pares de bases) se pueden incorporar directamente en un cebador de PCR, ya sea internamente, o en el extremo 5'.
- Si el ADN molde es lineal y el sitio de inserción deseado no está al final del molde, entonces el gen completo (más inserción) se puede producir usando PCR asimétrica o PCR superpuesta (es decir, mostrada anteriormente).
.png)
Figura 3.3.15: Mutagénesis por inserción
- Se pueden lograr grandes inserciones usando un molde (la inserción deseada) para PCR con los cebadores que tienen secuencias 5' que son complementarias a la región de inserción en el gen deseado:
.png)
Figura 3.3.16: Inserción grande
Mutágenesia “aleatoria” con PCR
- El protocolo de PCR se puede modificar para introducir mutaciones en posiciones aleatorias en el ADN diana.
- El principio detrás de la mutagénesis es la incorporación errónea de bases en posiciones “aleatorias”.
- La incorporación errónea por la polimerasa Taq, por ejemplo, se puede lograr agregando Mn 2+ al tampón de reacción, y disminuyendo la concentración de uno de los cuatro dNTP.
- En los sitios de la plantilla donde se debe incorporar la base reducida, habrá una mayor probabilidad de incorporación errónea.
- Así, la elección de la base con concentración disminuida determina los sitios en el molde que potencialmente serán mutados.
- La base mal incorporada es más o menos aleatoria.
- La concentración ideal de Mn 2+ a agregar varía entre 0.1 a 0.5 mM y se determina empíricamente. Las concentraciones relativas de bases es de 1 mM para cada base, excepto la base reducida, que normalmente está presente en una relación 1:5 o 1:10 (es decir, 0.2 a 0.1 mM).