
3.5: Secuenciación de proteínas, mapeo de péptidos, genes sintéticos
- Page ID
- 57249

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
Históricamente, algunos estados de enfermedad importantes fueron identificados como causados por la falta de una proteína importante, o la presencia de una forma mutada disfuncional de una proteína.
- Por ejemplo, se encontró que la diabetes, los tipos de enanismo y hemofilia se debían a deficiencias en insulina, hormona del crecimiento y factor VIII de coagulación, respectivamente.
Estas enfermedades podrían tratarse inyectando dosis suplementarias de preparaciones purificadas, o parcialmente purificadas, de estas proteínas.
- Estas proteínas se aislaron de materiales naturales, por ejemplo porcino (insulina), pituitaria de cadáver humano (hormona de crecimiento humana) o fracciones de sangre agrupadas de donantes normales (factor VIII).
- En la mayoría de los casos, aunque la proteína se encontrara en un suministro relativamente abundante, el costo de producción fue sustancial.
La mayoría de las veces, las propiedades bioactivas interesantes se asociaron con proteínas que solo podían aislarse en cuantidades diminutas (por ejemplo, el activador del plasminógeno tisular proteico que disuelve el coágulo sanguíneo).
Además, las proteínas no humanas generaban típicamente una respuesta inmune cuando se inyectaban en humanos, por lo que la forma humana de una proteína era la única forma útil.
- Si la proteína no estuviera fácilmente disponible a partir de sangre u orina, resultaría poco práctico obtener el material de partida adecuado para su producción.
- Desafortunadamente, si el material se derivaba de fuentes humanas, existía la posibilidad de propagación de enfermedades humanas (por ejemplo, la hepatitis y el virus del SIDA).
Si la información genética de estas proteínas pudiera aislarse, y luego transcribirse y traducirse en un sistema biológico fácilmente escalable, podrían obtenerse cantidades potencialmente grandes de proteína -y ojalá, de manera relativamente económica.
Con el desarrollo de la “biología molecular”, i.e.
- la estructura del ADN,
- la elucidación del código genético,
- la identificación de promotores transcripcionales y sitios de unión a ribosomas,
- el aislamiento de las endonuclases de restricción,
- la identificación del origen de la replicación del ADN
- el desarrollo de plásmidos con marcadores seleccionables, y
- el cultivo de E. coli,
existía la posibilidad a mediados de la década de 1970 de juntarlo todo y producir cantidades relativamente grandes de cualquier proteína humana para uso terapéutico.
¿Cómo se llevaría a cabo el proceso de producir grandes cantidades de alguna proteína humana importante? (es decir, purificación de proteínas)
El punto de partida es típicamente un ensayo para una funcionalidad de interés. Por ejemplo, podemos tener un hemofílico cuya sangre no coagule. No obstante, encontramos que si tomamos una muestra de su sangre y le agregamos una pequeña cantidad de sangre de un individuo “normal”, la sangre del hemofílico ahora coagulará. Esta será la base de nuestro ensayo.
Mediante este ensayo, fraccionaremos la sangre normal usando diversos medios: precipitación química (con etanol, o sulfato amónico), y luego varios pasos de cromatografía líquida, etc.
- En el camino seguiremos hacia dónde va nuestra actividad coagulante.
- Ojalá, en algún momento no podamos fraccionarlo más y tendremos una proteína pura.
Una vez que tenemos una proteína pura podemos comenzar a caracterizarla con respecto a su secuencia de aminoácidos. A partir de ahí podemos finalmente obtener el gen para la proteína y expresarlo.
.png)
Figura 3.5.1: Producción de proteínas
Análisis de secuencia peptídica N-terminal
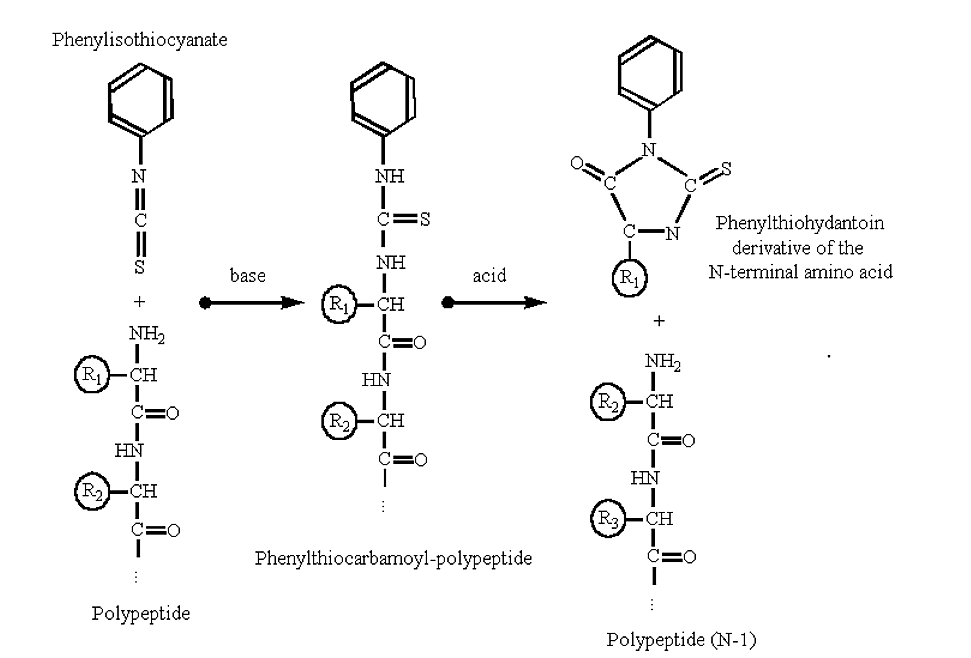
Los polipéptidos pueden secuenciarse a partir de su extremo amino mediante procedimientos automatizados basados en la reacción de degradación de Edman:
.png)
Figura 3.5.2: Degradación de Edman
- Tenga en cuenta que con la química de Edman solo se une y elimina el residuo N-terminal, el resto del polipéptido permanece intacto después de la reacción.
- El nuevo grupo amino terminal (anteriormente el segundo aminoácido en la cadena polipeptídica) ahora está disponible para otra ronda de reacciones. Así, el método puede ser automatizado.
- La cadena lateral de aminoácidos del derivado de feniltiohidantoína se puede identificar mediante cromatografía líquida. Los secuenciadores modernos de aminoácidos probablemente pueden secuenciar del orden de dos a tres docenas de ciclos (aminoácidos) de un polipéptido.
- Tenga en cuenta que la reacción requiere un grupo amino libre en el extremo N-terminal de la proteína. Si el residuo amino-terminal está metilado o formilado entonces la reacción no procederá (y se dice que el polipéptido tiene un N-terminal “bloqueado”).
Análisis de secuencia peptídica C-terminal
El análisis de la secuencia peptídica C-terminal no está tan bien desarrollado como el análisis amino terminal.
- El método generalmente hace uso de carboxipeptidasas no específicas.
- Las carboxipeptidasas hidrolizarán secuencialmente polipéptidos desde el extremo carboxi-terminal. El aminoácido liberado se puede identificar usando métodos de cromatografía líquida, y el polipéptido restante está disponible para reacciones adicionales.
- Varias carboxipeptidasas están disponibles, generalmente no son completamente inespecíficas (es decir, tienen ciertas preferencias):
|
Nombre |
Fuente |
Especificidad |
|
Carboxipeptidasa A |
Páncreas Bovino |
Aromáticos, alifáticos (hidrofóbicos) |
|
Carboxipeptidasa B |
Páncreas Cerdo |
Arginina, Lisina, Ornitina |
|
Carboxipeptidasa P |
Penicillium |
Generalmente no específico |
|
Carboxipeptidasa Y |
Levadura |
Aromáticos, alifáticos |
A veces, la elección de qué carboxipeptidasa usar se basa en la información de secuencia esperada. En este tipo de experimentos:
- se toman muestras en diferentes momentos durante la digestión
- los aminoácidos libres se separan de los polipéptidos
- los aminoácidos liberados se identifican mediante análisis de aminoácidos (cromatografía líquida).
El análisis C-terminal suele ser solo exacto para la identificación de la última media docena de residuos más o menos en un polipéptido.
Mapeo de péptidos
Uno de los problemas obvios con la secuenciación de proteínas es que incluso si el N-terminal no está “bloqueado” solo se puede obtener información de secuencia limitada a partir de un polipéptido intacto (es decir, solo alrededor de dos docenas del N-terminal y media docena del C-terminal).
¿Cómo se puede obtener la información de secuencia para todo el polipéptido?
Un método es el de mapeo de péptidos. El mapeo peptídico hace uso de escisiones proteolíticas del polipéptido para producir polipéptidos más pequeños. Estos polipéptidos más pequeños pueden aislarse entre sí y someterse a análisis de secuencia.
¿Cómo ordenamos las diferentes secuencias que obtenemos?
Una de las formas más fáciles es repetir el experimento, pero con una proteasa con una especificidad diferente, y de esta manera obtener información de secuencia solapada.
|
Nombre |
Fuente |
Especificidad |
|
quimotripsina |
Páncreas Bovino |
Escote después de Tyr, Phe y Trp; alguna escisión después de Leu, Met y Ala |
|
Bromelina |
Piña |
Escote después de Lys, Ala y Tyr |
|
Tripsina |
Páncreas Bovino |
Escote después de Arg, menos después de Lys |
|
Proteasa V8 |
Staphylococcus aureus |
Escote después de Glu, menos después de Asp |
.png)
Figura 3.5.3: Productos de escisión superpuestos
La información de secuencia superpuesta puede permitirle alinear los péptidos en el orden correcto y determinar la secuencia del polipéptido grande original (es decir, proteína).
Un problema que puede surgir se refiere a los residuos de cisteína y la naturaleza de cualquier puente disulfuro covalente en la proteína.
- Cualquier movilidades “peptídicas” (ya sea en cromatografía líquida o análisis PAGE) que se dividan en dos péptidos más pequeños después del tratamiento con un agente reductor (como b-ME) indican la presencia de un enlace disulfuro mediado por cisteína.
- Al secuenciar estos péptidos deben contener cada uno un residuo de cisteína. Si cada péptido tiene solo una cisteína, entonces la asignación del enlace disulfuro es inequívoca.
.png)
Figura 3.5.4: Residuos de cisteína en productos de escisión
Información genética correspondiente
Una vez que tenemos información parcial o completa de la secuencia peptídica podemos comenzar a identificar y aislar la información genética correspondiente. Este es el objetivo principal. Una vez que tenemos la información genética correspondiente, puede ser posible producir cantidades relativamente grandes del polipéptido deseado.
Traducción posterior
Como conocemos el código genético, podemos volver a traducir cualquier secuencia polipeptídica en una secuencia genética correspondiente.
- Así, a partir de la secuencia de aminoácidos podríamos sintetizar un gen artificial que codificaría para la proteína de interés.
- Dado que muchos aminoácidos están codificados por más de un codón, existe una ambigüedad potencial con respecto a la secuencia genética exacta original.
|
Aminoácido |
Número de Codones |
|
Met, Trp |
1 |
|
Phe, Tyr, Su, Gln, Asn, Lys, Asp, Glu, Cys |
2 |
|
Ile |
3 |
|
Val, Pro, Thr, Ala, Gly |
4 |
|
Leu, Arg, Ser |
6 |
Sin embargo, asegurarse de que volvemos a traducir de tal manera que se duplique fielmente la secuencia genética original puede no ser crítico; una secuencia proteica correcta es el objetivo general.
De hecho, si estamos intentando expresar la proteína en otro organismo (digamos expresando un gen de mamífero en un sistema bacteriano) en realidad podemos preferir elegir un sesgo de codón apropiado para el organismo huésped de expresión.
Los genes sintéticos para proteínas pequeñas son una forma razonable de proceder; esta es una forma en la que la insulina humana se ha expresado en sistemas bacterianos.
- Sin embargo, la síntesis automatizada de oligonucleótidos de ADN es práctica para longitudes de polímero de aproximadamente 60-90 bases o menos (aproximadamente 20-30 aminoácidos).
- “Además, el método de construcción de genes sintéticos normalmente requiere oligonucleótidos complementarios solapantes (para ser ligados en un “" casete "” de gen de ADN dúplex único).”
Por lo tanto, se requieren muchos oligonucleótidos incluso para un solo gen sintético pequeño.
.png)
Figura 3.5.5: Construcción génica sintética
Una forma de mejorar el método anterior de construcción de genes sintéticos es con un enfoque de PCR directa. Este método no utiliza ligasa, ni siquiera oligonucleótidos que se topan entre sí. En cambio, con este método se utilizan simultáneamente muchos (~100) oligonucleótidos superpuestos diferentes en una reacción de PCR. Su complementariedad de secuencia se puede representar de la siguiente manera:

Es posible que el conjunto completo de oligonucleótidos no se alinee para dar el gen completo, pero eso está bien. Haremos múltiples rondas de PCR con la idea de que algunos oligo complementarios se hibridarán y se extenderán y conducirán, poco a poco, a la construcción de un gen sintético contiguo:
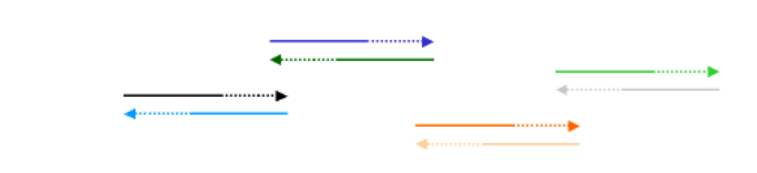
En el siguiente ciclo de PCR, algunos de estos fragmentos extendidos se hibridarán con otros:
.png)
Estos se extenderán a través de la PCR y pueden continuar hibridando con otros fragmentos de PCR más grandes. Eventualmente, se construirá todo el gen. Sin embargo, dado que la eficiencia de construcción del gen de longitud completa probablemente no va a ser muy buena, necesitamos realizar un experimento de PCR posterior para amplificar el gen de longitud completa (usando cebadores externos). Las características principales de este método se resumen de la siguiente manera:
- Muchos (hasta 1-2 cientos) oligo superpuestos se combinan en una sola reacción de PCR
- Los oligos están diseñados para ser lo más largos posible (~100 meros) con solapamiento limitado (~20 bases)
- El gen de longitud completa se construye en un experimento de PCR inicial (bajo rendimiento)
- Este gen de longitud completa se amplifica con un experimento típico de PCR posterior usando cebadores externos.